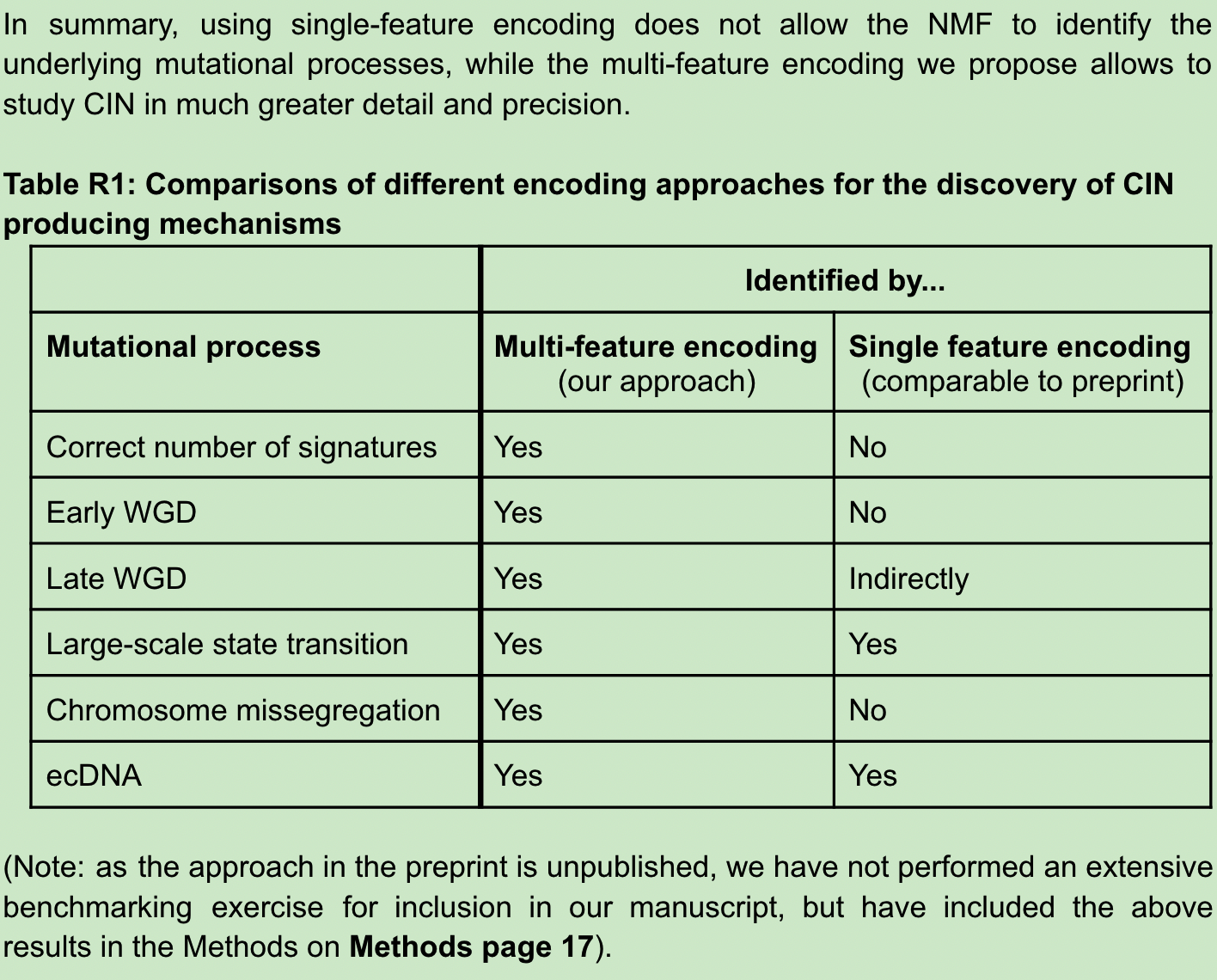
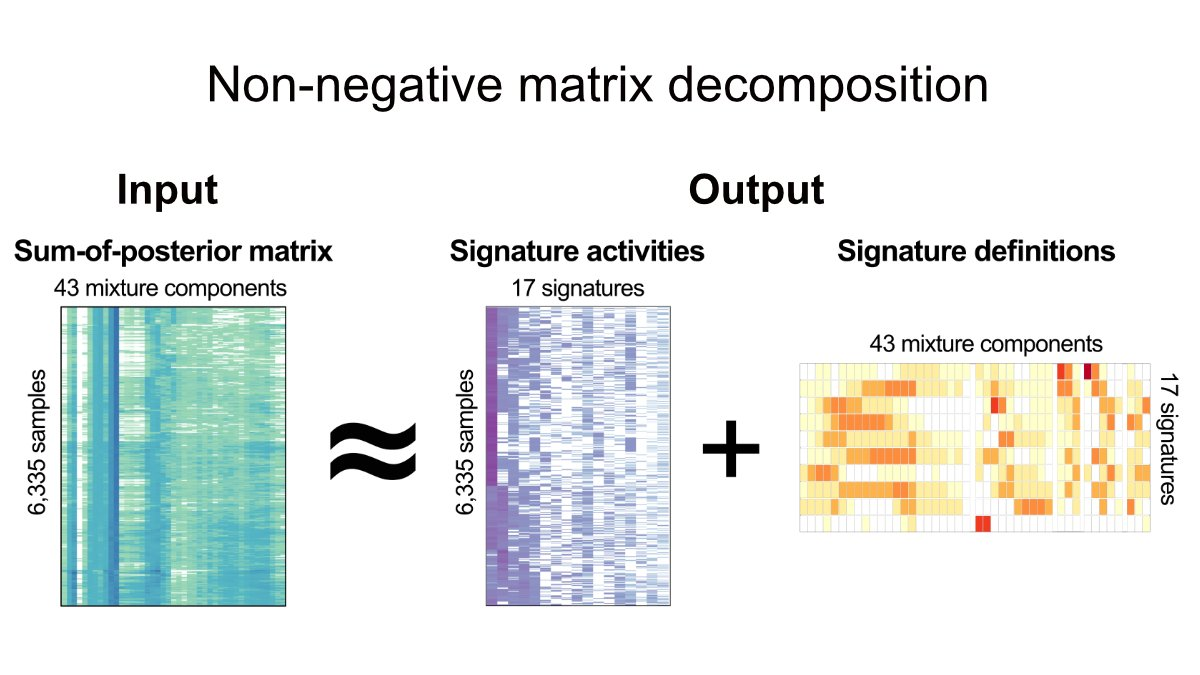
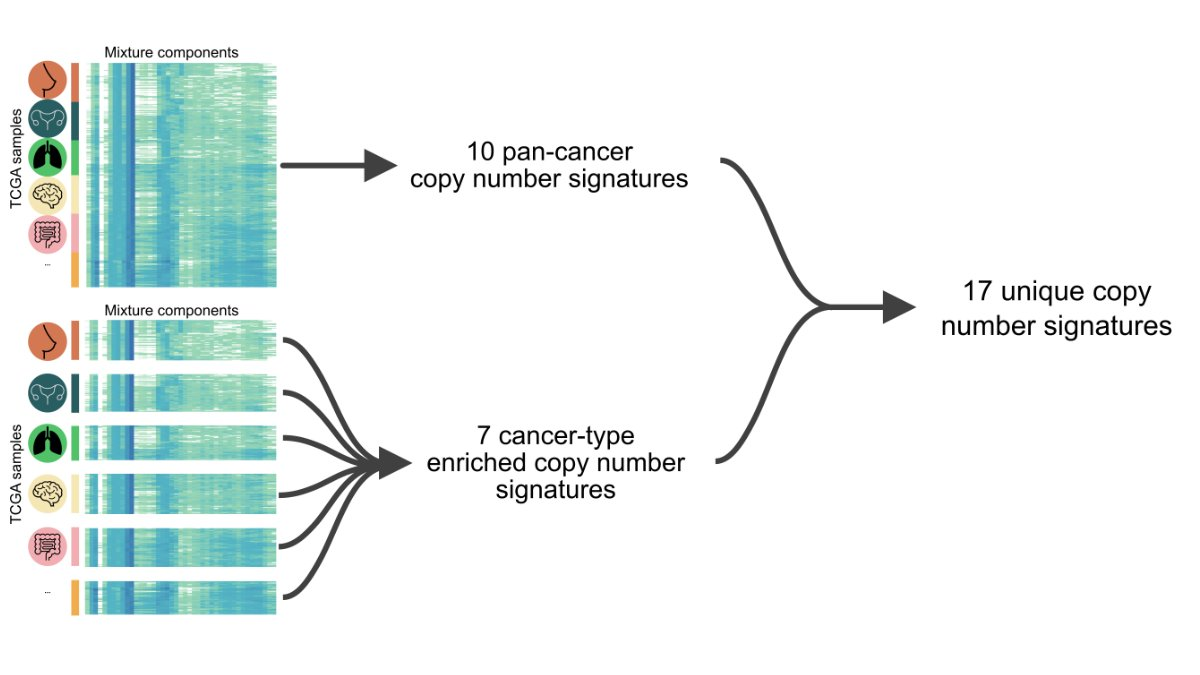
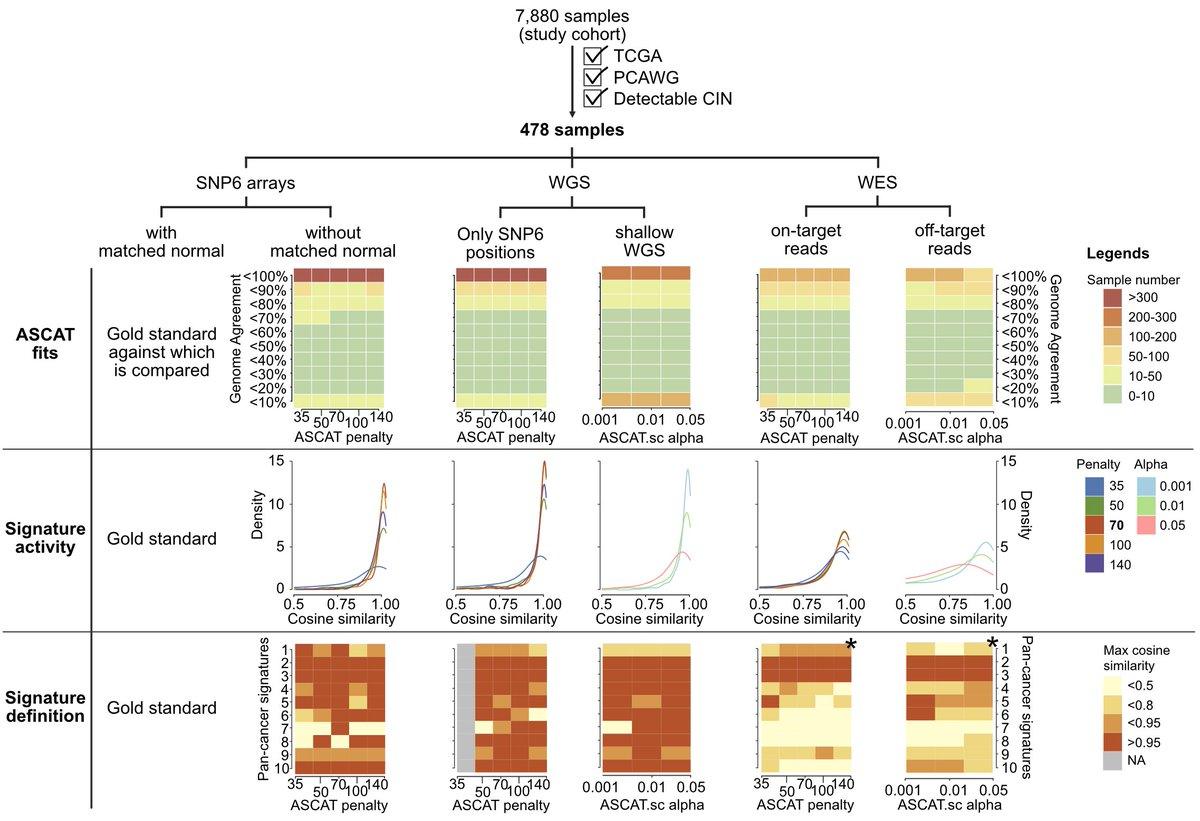
2022 年 6 月 15 日,Nature 在线发表了两篇染色体不稳定性泛癌研究。一篇题为:A pan-cancer compendium of chromosomal instability;另一篇题为:Signatures of copy number alterations in human cancer。 和我们常说的新闻「字儿越少事儿越大」一样,科学文献有一个潜在成立的标题定律,通常越重要越前沿的文章标题越短,比如 1953 年发表在 Nature 的 DNA 双螺旋结构 Molecular Structure of Nucleic Acids。 在肿瘤研究的很多方向上,如今我们只能通过不断增加限制条件和定语完成发表。所以如今看到10个单词以内的研究,不妨都可以多看两眼。这在我自己感兴趣的方向上,Nature一下online了两篇,那还了得。这此,我们先聊第一篇,剩下一篇有机会再说。 基因组(染色体)不稳定性作为肿瘤 20 年来人们已知的 hallmarks 之一,无论是单一癌种还是泛癌种,TCGA / PCAWG 前前后后都发过很多篇,如今用这些数据还能发表在 Nature 杂志上,而且还可以称之为泛癌种染色体不稳定性纲要(compendium),想必是有点东西。 而让我真正想仔细看看这篇文章的另一个原因是它波折的发表经历。文章的投稿时间是 2020 年 7 月 31 日,Nature接受时间是 2022 年 4 月 21 日,正式在线时间为 2022 年 6 月 15 日。一次投稿接近两年的修改时间,对于一篇没有湿试验的paper 来说,和 review 之间的拉扯不可谓不长。 前后经历了四轮修改的结果是文章一共68个figure,60页附件,90页Peer Review File。今天,我们就一起来学习这篇文献,同时试图聊聊其发表背后可能经历的波折和能学习到的经验。 以下几点是我在阅读之后提炼的部分文献学习要点,你在阅读原文时可以作为参考。 染色体不稳定性(CIN,chromosomal instability)对于肿瘤具有非常复杂的影响,包括驱动基因的丢失或扩增、大片段重排、染色体外 DNA(ecDNA)、微核形成和先天免疫信号的激活等等。这导致其了与肿瘤的分期、转移、不良预后和耐药等均有关联。 造成 CIN 的原因也是多种多样的,其中包括有丝分裂错误、复制压力、同源重组缺陷(HRD)、端粒异常和断裂-融合-桥周期(Breakage-fusion-bridge cycle)等等。 由于这些原因和后果的多样性,在目前的研究中,CIN 通常被当作一个综合事件来看待。 CIN 的测量描述,通常来说,或者是将肿瘤分为高 CIN 或低 CIN 两类,或者局限于如 HRD 这一单一病因,有或者局限于如染色体臂变化这类特定的基因组特征,再或者只能对特定的癌症类型进行量化。 目前还没有一个系统性的框架来全面描述泛癌症的 CIN 的多样性、程度和起源,也没有定义肿瘤内不同类型的 CIN 与临床表型的关系。(嗯,文章开头提到的两篇文章就都是在回答这个问题)。 为了解决上述提到的问题,作者首先使用来自癌症基因组图谱 (TCGA) 的SNP6 array数据,获得了 33 种肿瘤类型的 7880 个高质量绝对拷贝数图谱。使用的软件和筛选流程见下图。 作者使用自己团队 2018 年发表在 Nat. Genet. 的一项卵巢癌 CIN 研究框架,对 6,335 个全基因组拷贝数结果计算了先前已经证明有效的五个拷贝数特征,这些特征可以代表拷贝数的变化模式(CIN 不同潜在原因)。 这五个特征包括: 不理解的可以看原文method详细说明或者下面的示意图。 关于为什么没有把一些更容易理解的指标纳入考虑,作者也在附件方法中也给出了一些解释。 比如,排除绝对的拷贝数值是因为,多个拷贝数变化事件可能发生在同一基因组位置,因此该段的绝对拷贝数可能因各种变异事件的顺序而有很大的不同。 例如在全基因组复制 WGD 的背景下,一个单拷贝丢失导致拷贝数为 3,但在非全基因组复制的背景下则为 1,尽管它们可能是由导致缺失的同一突变过程引起的,但是会有两个不同的特征。 因为染色体倍性会对很多生物学特性有特殊的影响,不使用绝对数值作为特征可以纠正倍性的影响,避免出现在不同倍性背景下编码相同原因的冗余特征。(多说一句,其实这一特征在文章开头提到的另一篇同时发表的Nature文章是用到的) 对于不考虑杂合性缺失(LOH)状态,是因为作者大多数 LOH 事件是所谓的拷贝丢失事件,可以通过拷贝数变化来检测。在文章使用的数据集中,他们发现整个 TCGA 中的 LOH 事件中位长度为 3.6Mb,这表明大多数事件可以通过 SNP6 技术或者 WGS 检测到。但是随着科学研究越来越多的用到了 shallow WGS 和单细胞测序,它们尚不能提供准确的等位基因特异性分辨率,因此专门的 LOH 特征可能导致这个方法的应用受限。(再多说一句,其实这一特征在文章开头提到的另一篇同时发表的Nature文章也是用到的) 看到这里,我会有一个疑问:基于测序数据的这几个指标是否可以用来描述和区分生物学中常见的CIN相关事件。 如下图所示,这里作者也列出了经过充分研究后上述五个具体的拷贝数特征能够捕获哪些已知的拷贝数pattern。而正是这些这些促使作者选择了这五个基本特征来模拟已知的pattern,并有可能区分其突变过程。 有了这 5 个特征之后,作者应用混合模型来定义每个队列范围内特征分布的不同成分,最终在 5 个特征中共确定了 43 个混合成分。其中片段长度包含了 22 个成分,拷贝数变化包含了 10 个成分,如下图所示。 从理论上讲,这些43个成分就代表了定义 CIN 过程的基本模块。然后就可以利用这些混合成分对每个肿瘤基因组进行编码,通过概率计算将拷贝数事件分配给这些成分,形成一个 6,335×43 的矩阵。然后,应用非负矩阵因式分解的来识别拷贝数 signature。 作者首先使用完整矩阵,发现了 10 个泛癌症拷贝数 signature。然后使用矩阵子集,代表至少有 100 个样本的单个癌症类型进行分析,首先找到了 128 个癌种特异性 signature。 随后通过三步过滤,来避免 signature 冗余。 最终找到了 17 个拷贝数 signature(10 个泛癌和 7 个癌种特异性),在下图中你可以看出这 17 个 signature 中不同拷贝数特征如片段长度和断点数等的 43 个成分的权重。 使用线性组合分解计算它们的活跃度,最终就可以得到一个泛癌的 17 个拷贝数 signature 和它们在 33 种癌症类型的样本活跃度。 这篇文章的亮点在于不是讨论CIN之后会怎么样,而是解释哪些原因导致了CIN。因此就需要将 17 个 CIN 特征与产生 CNA 的推测原因联系起来。(思考题,如何用已知解释未知并且让人觉得可信,是生信最大的难点)。 关联的整体思路: 总体来说,为了完成这一步,如下图红框所示,他们用到了目前几乎所有可用的公共数据。 两个患者队列的数据及其临床数据 五种突变特征 14 个分子特征 11 个 DNA 修复特异性特征 为了将 signature 活性和突变基因关联起来,作者进行了如下的筛选。首先通过 driver gene 的既往研究挑选出了 200 个肿瘤相关基因,根据 reactoma 数据库筛选出 1330 个 CIN 相关通路蛋白。对筛选后的 1179 个基因按照有无突变分为两组检测和17个CIN signature的相关性,找到788个至少和1个signature相关的基因。在这788个基因中通过qvalue和effect size进行过滤,保留了32个和signature高度相关的高可信度基因。 这32个基因是 AKT1, BRCA1, BRCA2, CCND1, CDK4, CDKN2C, CIC, CUL1, ERBB2, ERBB3, ERCC2, FBXW7, H3F3A, IDH1, IDH2, MAPK1, MYC, NFE2L2, RAC1, RPL22, PBRM1, PCBP1, PIK3R2, PMS1, PPP2R1A, PSMA4, SPOP, SOS1, SOX9, TP53, U2AF1, VHL。作者研究了围绕这些基因的文献,并整理了关于它们如何影响基因组稳定性的信息。 这些基因突变和拷贝数signature存在着如下关系 再上图你可以看到一些基因被红线划掉了。这是为什么呢? 对于如上图所示显示出扩增模式的四个特征(CX8、CX9、CX11、CX13),作者研究了它们与染色体外DNA扩增子的关系。由于致癌基因容易被ecDNA扩增,作者假设通过ecDNA扩增的基因可能是产生这些扩增子过程的结果,因此可能与突变过程没有因果关系。用相关研究文章的数据测试了在ecDNA扩增子中扩增的高置信度基因的特征活性的变化情况,对于扩增相关signature中的每一个高置信度基因检验带有该基因扩增子的样本是否比没有扩增子样本具有明显的signature活性。因此额外删除了几个用于假设CIN成因的基因。 根据signature和CIN相关联的病因假设逻辑如下图。 如果一个signature没有和它相关联的基因,那就没办法做出假设。如果一个signature不包括扩增但是有相关联的基因,查找这个基因和CIN相关的机制,如果有已知的机制就给出假设;如果一个signature包含了扩增且有基因关联,首先判断这个基因是否在ecDNA中也富集,如果富集了就假定不存在关联。然后还有剩余基因的,根据和CIN的相关功能进行假设。利用这样的分析共得到了11个signature可能的假定成因。 有了这些可能原因之后,再尽可能多的利用外部数据进行验证,给出打分。 如果高置信度的基因将某一特征与已知的CIN类型联系起来,加两颗星。如果有额外的数据支持该假定的原因,我们就增加第三颗星。 最终,八个signature(CX1-CX6,CX8,CX11)获得了三颗星。三个signature(CX10, CX14, CX17),即至少有一个高可信度基因与已知的CIN类型相关,但没有额外的数据支持。三个signature(CX9、CX13、CX16)被授予一颗星,这些特征没有高可信度基因,但一些额外数据验证使作者能够提出一个推测的病因。三个特征(CX7、CX12、CX15)为零星。 这里可以看一个具体的实例,比如在CX3中,本身的突变特征提出了假设,它具有LST可能和HRD相关;同时一个拷贝数的改变显示它可能是LOH事件和tandem duplications。 基于此,根据关联基因发现以下这些基因的功能确实和CIN的功能相关,首先给两颗星。那么是否有其它数据的支持呢?可以看到各种其它 HRD 相关的signature都存在,且还有生存数据的支持,最终三颗星。 经过这样一通复杂的验证,最后得到了下图这样一个核心结果。关于详细成因的解释还是看原文吧。 染色体异常与七个不同的特征有关,表明许多潜在的原因是这些复杂重排的基础。复制压力与八个signature相关,突出表明它是CIN的一个主要来源。不同的特征显示出在WGD之前(CX1、CX2、CX7和CX15)或WGD之后(CX3、CX5、CX6、CX8、CX9、CX13和CX17)发生的偏向,显示了WGD事件在调节CIN方面的重要性。APOBEC突变和kataegis特征与六个signature相关,强调了这些是CIN的一个共同特征。 有了原因的解释,这些signature可以用来做什么呢? 上述成因的探索结果暗示了典型的癌症相关同路是CIN的一些主要驱动因素。而这些途径中的许多基因都是靶向治疗的重点。 因此,鉴于这些signature可以很容易地在病人肿瘤中测量,作者接下来探讨了它们在治疗反应预测和药物靶点识别方面的效用。 作者整合了来自297个癌症细胞系的数据,结合通过CRISPR-Cas9或RNAi筛查确定的基因重要性以及对药物扰动的响应相关联,来发现相应的生物标记物和新的药物靶点,进而评估signature活性和基因以及药物敏感性之间的相关性。 分析结果如下图所示,作者确定了40个基因的拷贝数signature与靶点的遗传和药物扰动都有明显的相关性。 在这些基因中,CX5是HR相关的一个特征,预测了通过抑制PARP1对奥拉帕里的反应,又因为这一特征也与RNAi敲除PARP1相关,因此可能代表了一种生物标志物对常规蛋白功能的抑制而不是PARP的捕获。 CX9(复制压力有关)与针对参与主要有丝分裂途径的基因(EGFR、JAK1、MET、PRKCA和PIK3CA)的多种激酶抑制剂的反应相关,这表明多激酶抑制剂的方法可能适合复制压力相关的肿瘤。 同时,从CRISPR和RNAi扰动筛选中,找到了104个具有可药用结构的靶基因,这些基因目前在临床上没有靶向治疗方法。这些代表了预测的合成致死药物靶点,其中49个有证据表明与CIN相关的机制有牵连(如上图所示)。 三种同源重组受(impaired homologous recombination,IHR)相关signature表明了CIN复杂性增加的一种模式。 即单独的IHR会产生CX2,它代表了串联复制的小拷贝数变化特征。IHR加上复制压力会导致CX5,它涉及较大的CNA。最后,IHR加上复制压力、NER受损和DNA损伤信号受损会产生CX3,其最大的CNAs与杂合度的丧失密切相关。不过研究结果没有说明这些不同层次的复杂性是以递进的方式发展的,还是由独立的过程发展。 HR和NER的损伤已知对铂化疗的敏感,鉴于只有CX3与NER的破坏有关,作者假设IHR特征可能表现出不同的铂敏感性预测能力。 由于卵巢癌患者经常接受以铂为基础的化疗,于是用Cox模型测试了三个特征预测总生存的能力,从而推断铂敏感性。可以看到,CX2与铂敏感性没有关系,CX5可以预测耐药性,CX3可以预测敏感性。 鉴于这些IHR特征能够预测铂化疗反应,作者进一步假设是否可以结合这些特征提供更好的铂敏感性预测指标呢? 由于CX2不具有预测性,作者首先将其作为捕捉非预测性IHR相关基因组变化的参考,并要求预测性的CX3活性超过它才有可能赋予敏感性,即如果CX3活性大于CX2,则预测敏感性。 如下图所示,这种可解释的分类器能够区分BRCA1胚系突变的卵巢癌队列、TCGA队列中的卵巢癌队列和独立验证队列和也常规使用基铂化疗的食管癌队列的总生存期。其实此前也有不少基于机器学习的复杂模型进行预测,但是作者测试发现这种简单的分类也不必之前需要更多信息的方法差。 以上就是这篇文章的主要内容,因为60多幅图和60页的附件实在是太多了,我的梳理能力也有限,如果你读到这里产生了其他疑问还是得自己再去看看原文。 接下来我们聊点或许能让你感兴趣的。 正如文章开头所讲,这篇paper从投稿到online用了接近两年的时间,对于一篇没有湿试验的 paper 来说和 review 的拉扯不可谓不长。 经历的四轮大修,我仅仅是读了一遍他们 92 页的交锋记录就感觉血压上来了好几次。如标题所言,2022年用TCGA数据研究染色体不稳定性还能发Nature,凭什么? 这不是我的疑问,而是几位审稿人的质疑。 如果你真的想提高自己的科研思维,我以为或许读Peer Review File比读原文更有价值。 首先,值得祝贺的一点是这篇文章一上来没有被editor直接拒掉,好在顺利到了三位审稿人手中。 通过阅读 Peer Review File,可以看到第一次投稿 review 后,三个人里,第一个提出了 8 大问题(Specific points)和 5 小问题(minor);第二个提出了 6 点大修和 4 点小修;第三个则是直接给出了 5 点主要问题(major issues)。 好在,这一次应该又是editor手下留情,给了作者大修的机会。以下是我觉得比较有趣的部分内容。 他的第一次的评论很不客气,说白了就是拒稿的意思。他提到: 而在8个Specific points中,审稿人主要质疑了如下几个问题。 评论如下图。从第二个审稿人的回复中,可以看出来第一版的投稿作者似乎只解释了两个在所有癌种里最常见的signature,且这些对应的生物学过程目前已经研究的比较多,认为应该关注更多的其它signature解读。 他提到的主要质疑包括 在第一次掰头的时候,看起来评价的很克制,但其实他的质疑最大,也造成了后面几轮引入了第四位审稿人。 第三个审稿人认为尽管CNV特征可以为了解CIN提供信息的想法是有希望的,但作者所描述的方法不够新颖或信息量不够,而且这些发现缺乏坚实的支持证据。 他的主要质疑包括如下几个问题: 我们这里小结一下,几个人最关键的就是两个终极拷问: 投稿的时候大家都有经验,关于审稿人的问题需要逐个回复,感兴趣的话就自己读读,这里只说几个最关键的修改。 作者的原话是:这次修订包括重大改进和额外结果,我们完全改变了文章的内容。那翻译一下就是重写了一篇。 虽然我们没办法看到第一版的文章,但是从这几个修改可以看出来,作者第一次敢直接投Nature也是挺勇的,大不了就是做一个被Nature拒稿的人。 关于稳健性测试这一块,其实最后的文章里作者在附件和附图中有了大量的分析来证实。为了验证这个 signature 模型的鲁棒性,作者在 TCGA 中筛选出了有 PCAWG WGS 数据的 478 个样本,对 WGS 数据进行基于 SNP6 芯片的下采样和 shallow WGS 下采样,将 WES 数据区分为 on-targets 和 off-targest,进行一致性评估。 还通过在人类基因组中引入拷贝数变化,模拟了如下五种染色体不稳定的活动: 然后再用自己的方法验证是否是不是能把这些内容还原出来。 临床应用价值这部分第一版看起来就是在fig1引入了一个CIN和生存的相关性,这次作者的回复是认怂,那我们就把这部分内容删掉。然后你要临床价值,那我就给你好好做做临床价值,才有了上文看到了药物预测和化疗敏感性预测。 作者一顿回复之后,迎来了第二波攻击。 第一个略被说服,重新提出了 6 个小问题 第二个人也表示肯定,但是表示因为你有重写了一篇文章,那我再根据你新写的文章接着追加 6 个小问题。 在前面两个审稿人态度转变之后,第三个审稿人开始发难了。 你可以感受一下他大概说了什么,以下为大致翻译。 自第一次提交以来,作者做了大量的工作,包括对另外几个人类癌症队列和细胞系模型的分析。他们在BRCA1突变的卵巢癌开发了一个基于CIN特征的分类器和一个预测铂化疗的模型。这是一个有趣的观察,但是由于WGS的病例数量较少,无法断定HRDetect是否能实现这一目标(难道他是HRDetect方法的作者么?)。 作者还比较了不同的测序平台,以评估他们的CIN特征方法的稳健性,但是这些比较是基于下采样的WGS/WES数据,不清楚当他们的方法应用于原始WGS/WES数据时,是否仍有相同程度的稳健性(这是不满意你没花钱测新的样本?)。 尽管做了大量的额外工作,我们认为新版本的主要结论并不令人信服。我们认为这篇文章存在着严重的方法学问题,而且我们发现signature的原因解释是推测性的。因此,我们认为这个文章不适合在Nature上发表。 关于为什么有严重的方法学问题,为什么不适合发表,他写了很长内容很多个问题,其中三个问题如下: 这一波操作,想必作者已经多了很多信心,没有再重写一遍文章,「只是」增加了7个figure,6张表。 这里我们重点挑几个对于审稿人3相关问题的回答看看: 对于CIN signature 和 HRDdetect 比较对于BRCA1胚系突变患者铂化疗敏感性的问题,作者说HRDdetect只有三例可用样本,你不是说是因为样本少看不出效果么,那我就直接从正文里拿掉了,顺便补个刀,HRDetect算法在训练的时候是把BRCA1/2突变当作正向label使用的,我们也不指望它能在突变组内部再有什么作为。 对于审稿人质疑CX2和CX5可能并不意味着这些特征是特异性的由于同源重组受损造成,而是一个副产物,作者里巧妙的使用CX2和CX5 CX3 这三个signature以及癌种在BRCA各种突变类型的样本之间做了一个多因素分析。证明,在多因素矫正后,CX2和CX5然后是显著在BRCA1突变的样本中有差异,因此表明CX2和CX5可能与BRCA突变状态直接相关,即是HR受损的结果。 对于审稿人质疑CX5与PARPi的敏感性有关但同时也与铂耐药有关互相矛盾时,作者则给出了如下答复。 这是一个有趣的观察(言外之意就是你看的好仔细),这确实似乎违反常识,因为公认铂敏感性也意味着 PARPi 敏感性。CX5似乎表明一些铂类耐药肿瘤也可能对 PARPi 敏感。这在文献中得到了一些支持,其中对铂耐药的胚系 BRCA 突变型卵巢癌对 PARPi 显示出敏感性。然而,我们认为我们没有足够的数据来进一步探讨这个问题,并没有在文章中强调它(我们的结果也有文献支持,你可以多查查文献)。 作者这一波回复之后,前两位审稿人已经表示可以了。 有趣的是,审稿人2甚至还回复了审稿人3的一些看法,顺便给作者出了主意,很可能是他也看不下去了吧。 压力来到第三位审稿人这里,这次他的评论可以看出来已经是「愤怒」了。 有趣的是,在这次评论中,这位review做了两件事情。他首先引用了一篇2021年发表在预印本的CNA signature文章,说这篇文章就没有犯作者的错误。其实,他引用的这篇文章正是开头我们提到的一起背靠背发表的另一篇文章。 第二点,为了证明作者使用的模型真的有问题,他甚至自己做了一个模拟研究,以证明所提出的方法存在问题。而解决这个问题的可能方案就是使用另一篇背靠背发表的文章。 事已至此,最终压力给到了editor这边,三轮之后三位审稿人二比一,但是第三位态度非常强硬,那就不得不动用第四位审稿人来当一次裁判进入最终的第四轮修改。 这一次作者的回复是四轮答复中最有趣的。 为了让第四位审稿人吃瓜吃的明白一些,作者首先解释了一下前面三轮审稿发生了哪些事情。以下为作者的心路历程。 在整体回复的最后,作者还不忘说了这么一句话: 随着时间的推移,我们与审稿人讨论的主题变得更加详细和技术,但是重要的是不要忽视更大的问题。我们这个研究将大量和全面的基因组数据合成到一个连贯的框架中来衡量染色体不稳定性。这一框架为加深对CIN的理解提供了重要的一步变化,该领域在过去几十年中只取得了零星的进展!(哈哈哈,你们差不多行了)。 大局观归大局观,作者在这一轮用两个主要问题的回答,算是彻底结束和第三位审稿人的讨论了。 审稿人3的第一个问题是引用了一篇他认为没有在方法学上犯错误的同类型文章。 那作者就用第二位审稿人出的模拟基因组的主意,进行了一次模拟比较,设置了五种不同的突变过程,而另一篇文章只给出了3个正确的结果。 至于原因,作者解释为由于单特征编码使用了绝对拷贝数,同一个signature将在不同倍性背景下被人为地分割,这也是他们在多特征编码中删除拷贝数的原因。CHR、WGD-early和WGD-late三个过程被多特征编码所捕获是因为它们能够在整个特征空间中代表CNA的许多生物学特性。例如早期和晚期的WGD尽管有相似的拷贝数变化数,但可以通过片段大小的特征以及每10MB的断点数量区分。(写到这里,不知道另一篇背靠背的文章作者怎么看呢) 审稿人3的第二个问题是自己模拟了一个分析,使用NMF未能重现输入的signature,进而证明方法有严重问题。 作者表示,审稿人的模拟并不是我们方法的有效模拟而是存在一个致命的设计缺陷,人为地导致NMF过程失败。所以又给出了一个正确模拟的例子验证了结果。 至此,三轮掰头结束,整个的审稿过程基本也就要结束了。 第四轮只有审稿人4发表了一些评论,认为作者已经很好的回答了之前几轮的问题,这是一篇会对相关领域产生有益补充的重要研究。而他也只是提出了一些文章表述上可能需要修改的问题。 回顾一下四次审稿过程的经历,可以总结出作者的文章经历了如下变动。 通过阅读四次大修的故事,也许你已经完全意识到了生物信息方法在公用数据中发表一篇Nature可能会经历哪些磨难,但不得不坦白讲,你我应该经历这种磨难都概率很小。 那明知如此,我为什么还有写上万字来记录这个过程呢?其实,我废了半天劲把90多页Peer Review File读完的真实用意,是想尝试搞清楚一个Nature级别的研究从执行到发表,故事性和逻辑性上会有怎样的提升。 幸运的是,在读完这折磨人的90多页文章修稿过程之后,我看到了通讯作者自己在社交媒体发布的文章主线介绍。也终于完成体会了,一个支离破碎修修补补科研项目,如何变成了一个严谨的激动人心的科学故事。 在本次推送的最后,你也来读读吧。以下内容来自于通讯作者的社交平台,我进行了大致翻译,大家重点体会此刻这个研究的呈现逻辑。括号里的内容是我的自行补充说明 多年来,我们一直在与CIN肿瘤作斗争。我们经常看到TP53突变在CIN中出现,但很少看到其他驱动因素。一段时间后,我们开始在混乱中看到了规律,并开始意识到这些特征代表了CIN的不同原因。这些可以帮助我们更好地理解CIN吗?(提出问题) 我们的第一次成功是在卵巢癌研究方面,在那里我们发现了如何对DNA拷贝数(CN)模式进行编码以允许因果机制的识别。这包括片段长度、断点数量等等。作者从浅层全基因组测序的高级别浆液性卵巢癌(HGSOC)病例中确定了拷贝数特征。(介绍背景) 此后,我们改进了之前的编码方式,如不再包括片段的绝对拷贝数。这似乎与直觉相反,但却出奇地有效。现在,不同倍性背景下的相同CIN类型被一个单一的模式所捕获,而不是许多模式,这使我们能够实现泛癌的分析。(介绍方法改进) 我们从以前的研究中知道,每个肿瘤都会有多种类型的CIN,导致基因组中出现多种模式的重叠。因此,我们使用贝叶斯版本的NMF,用我们的新编码解开这些模式。 我们在泛癌种中应用NMF确定了10个特征。然后将其单独应用于每个大的癌症类型中又确定了7个泛癌特征。这使我们总共得到了17个泛癌CIN signature。 我们通过在模拟基因组中正确识别5种已知的CIN原因的组合,来测试编码和特征识别的稳健性。这五种特征包括 mitotic errors, cytokinesis failure, endoreduplication, homologous recombination deficiency and ecDNA circularisation/amplification。 我们的特征在不同的基因组分析技术中也是稳健的。无论是SNP芯片,深层和浅层全基因组,以及外显子组测序。(第二次修改才增加的内容) 至此,我们有信心拥有了一个强大的17 CIN signature,我们必须攀登下一个高峰:了解这些特征背后的生物学。(递进,介绍第二部分解释生物学原因) 第一个挑战:由于NMF产生了稀疏的signature定义,我们需要使它们更容易解释。为了做到这一点,我们把来自输入矩阵、活跃矩阵和定义矩阵的信息结合起来,生成了一个解释矩阵,其中每个signature都是在每个分解成分中被定义的。(第三次修改才增加的内容) 这个矩阵使我们能够开始理解我们的signature,在许多情况下,编码模式已经暗示了一种机制(例如,大长度片段+每条染色体未发生断裂=有丝分裂错误造成的染色体错位)。 第二个挑战:将signature与有缺陷的途径联系起来。为了做到这一点,我们测试了有突变驱动基因的病人的signature活跃度是否明显较高。这些联系,加上上面的矩阵使我们能够为12个siganture 提出推测的原因。(第二次修改完善) 第三个挑战。为假设的原因找到支持性证据。为了做到这一点,我们整合了大量的数据,以验证完善我们的推测。(第二次修改补充) 攀登了这座巨大的解释之山后,我们看到了山顶上的风景。3个有丝分裂错误的信号;3个同源重组受损的信号;4个复制压力的信号;1个NHEJ受损的信号;1个PI3K介导的WGD(星级=证据的强度)。5个未知数。 当我们考虑这个观点时,我们意识到许多被确定的缺陷因果途径包含正在被治疗的驱动因素。CIN特征能否预测对这些疗法的反应?或甚至确定新的药物目标?(你没意识到啊,第一稿根本没想到这个) 为了探索这一点,我们将297个癌症细胞系的signature与来自CancerDepMap的全基因组CRISPR、RNAi和药物筛选数据结合起来。我们发现我们的signature可以预测对44种靶向治疗的反应和49个CIN相关的可药用基因的扰动评估。 鉴于这些令人兴奋的体外结果,我们想知道......我们是否也能预测患者对CIN癌症的主要治疗方法之一的反应:基于铂的化疗?(这也是被审稿人diss才加上的) 以前,HRD已被证明可以预测对铂化疗的敏感性。但现在,我们有办法利用3个IHR特征来分解 "HRD "表型。 为了测试这一点,我们把重点放在了常规使用铂化疗的卵巢癌上。使用总生存期作为敏感性的替代物,我们发现CX2没有预测性,CX5预测抗性,而CX3则是敏感性!这已经非常棒了。但如果我们把它们结合起来呢? 基于胚系BRCA1/2突变体可能有HR损伤,但不一定对铂化疗敏感的想法,我们想出了一个简单的分类规则,将 "背景" CIN(CX2)从预测性CIN(CX3)中剔除。一个简单的规则,如果CX3>CX2,则为敏感,否则为耐药。 这个简单的分类器非常强大,能够对统一使用铂治疗的患者群体进行剖析。这包括BRCA1胚系突变的卵巢癌,以及其它卵巢癌、食管癌和乳腺癌的队列。其性能也可与更复杂的方法相媲美。 多么好的旅程啊,我们的signature提供了一个了解CIN的窗口,并提供了一个框架来评估泛癌症的染色体不稳定性的程度、多样性和起源。(升华主题,上价值) 为了更快地将这项技术带给病人,我们还成立了TailorBio。这家科技生物初创公司拥有将这项技术转化为精准医疗平台的独家权利,该平台用于改善患者分层并开发新的靶向治疗。 下一个研究前沿是研究CIN的动态变化。我们正在开发技术,利用来自早期病变、KO细胞系和药物处理过的器官进行单细胞基因组分析,识别正在进行中的CIN。欢迎你和我们联系。(介绍正在做的事情,占领地盘顺便招人) 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱或者微信订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。写在前面
或许能学到的
关于染色体不稳定性
核心研究思路和方法
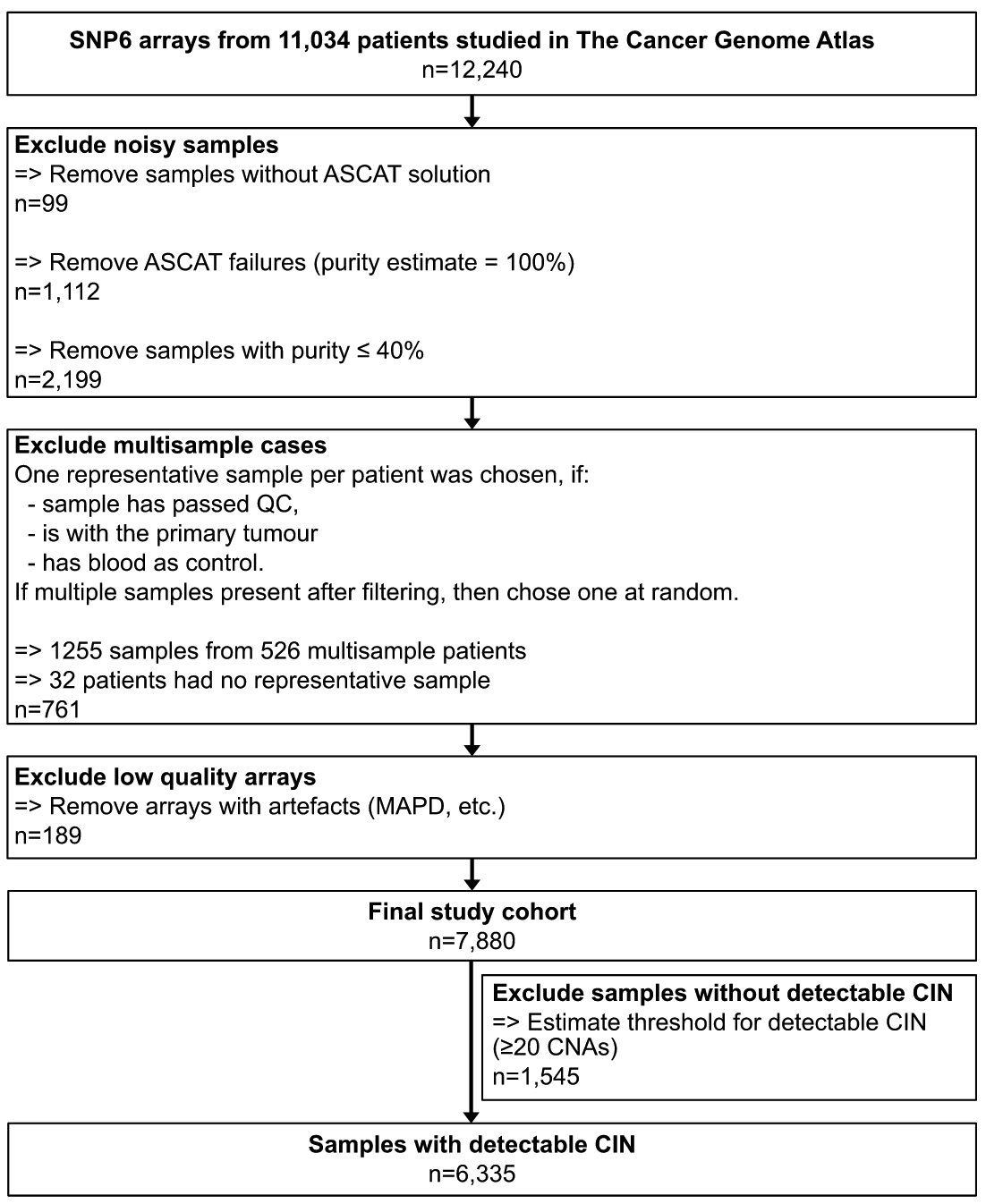
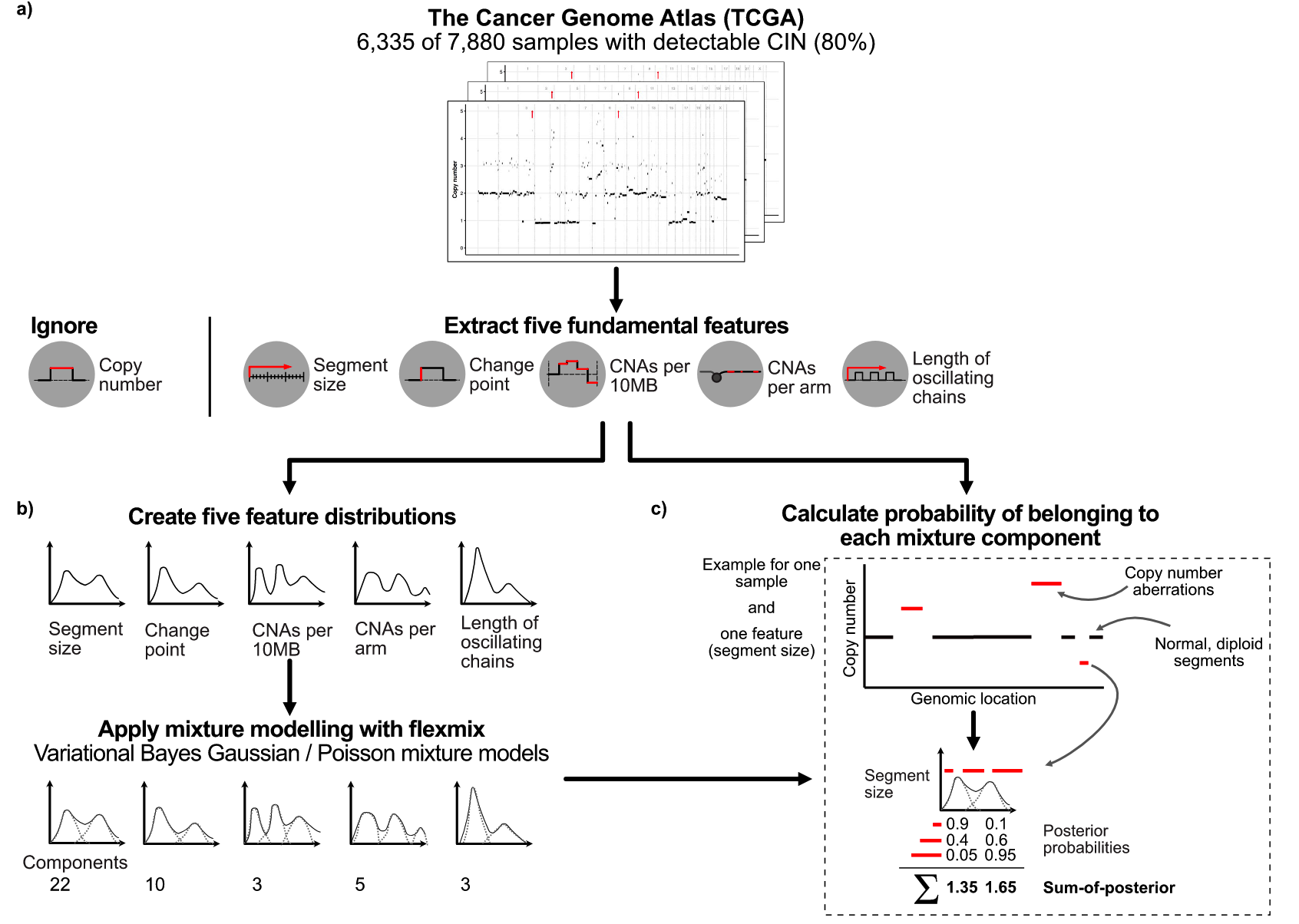
研究样本
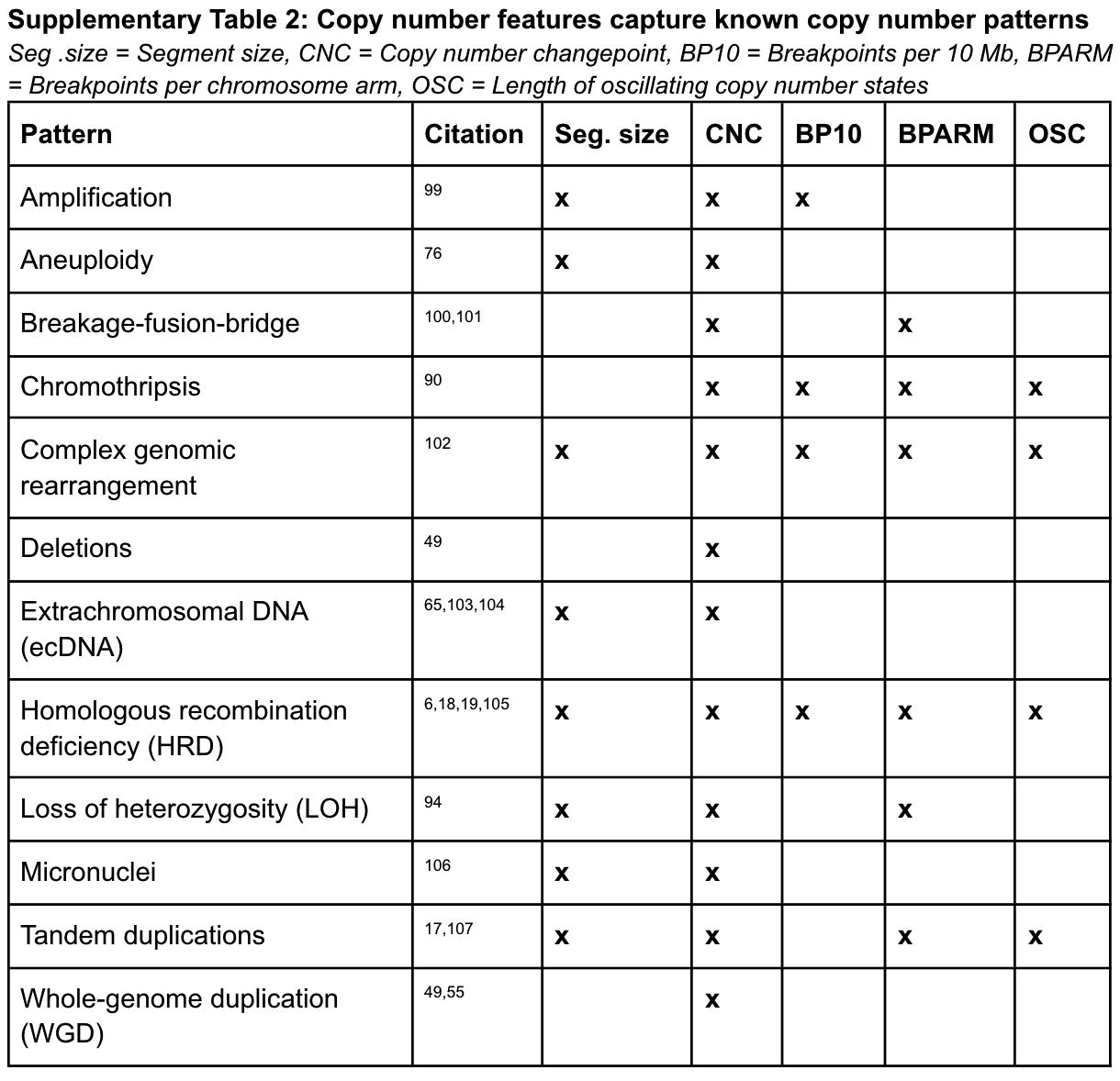
特征选择

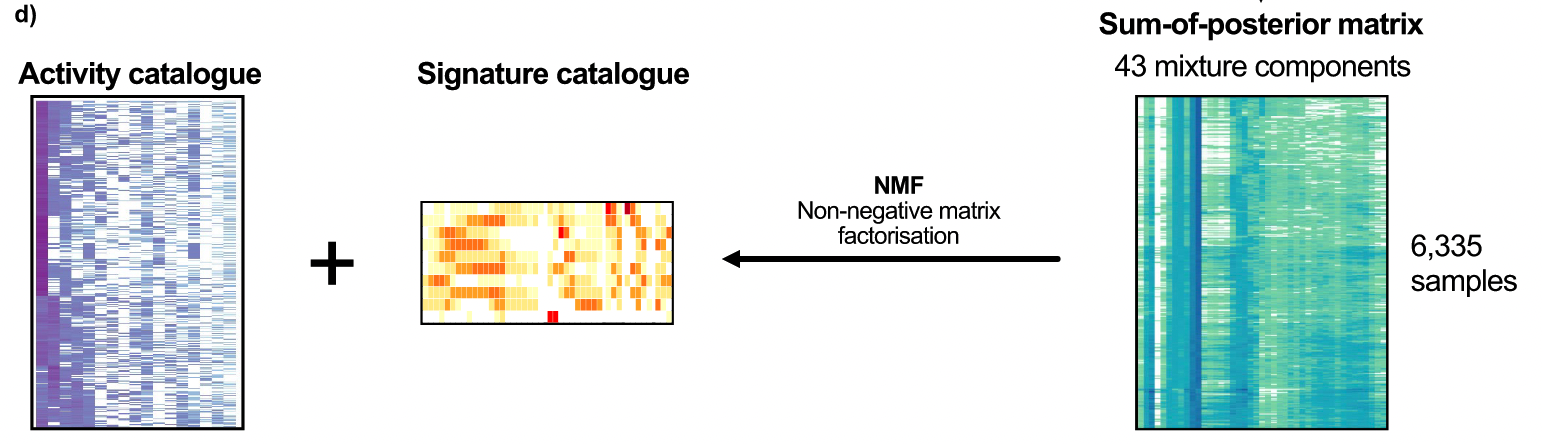
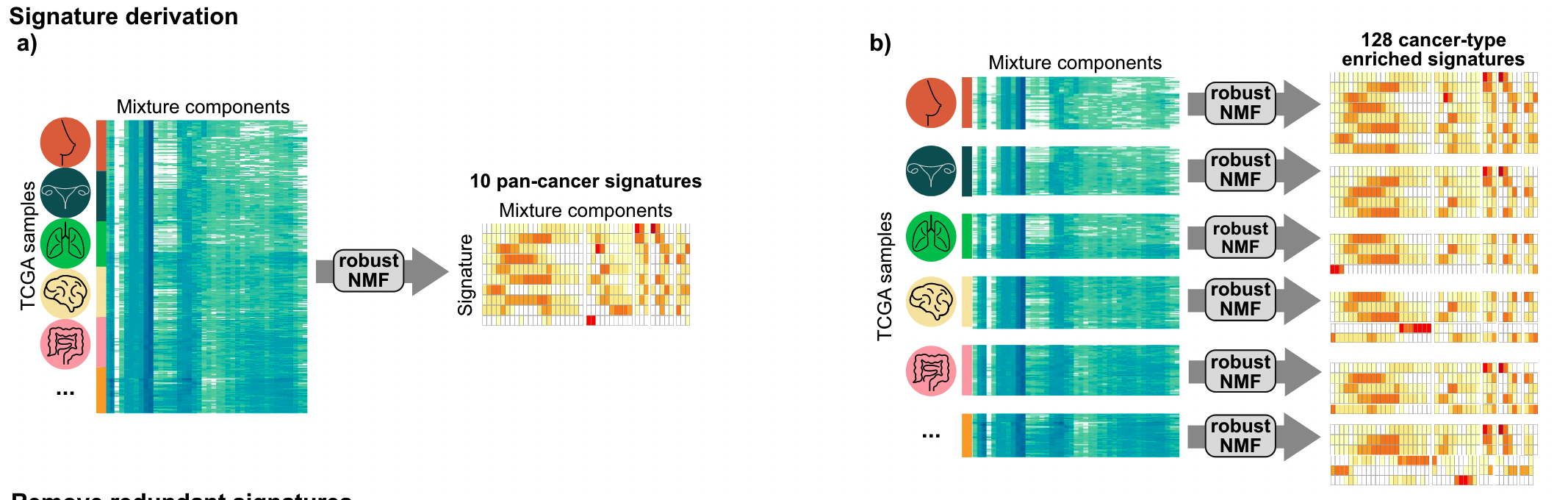
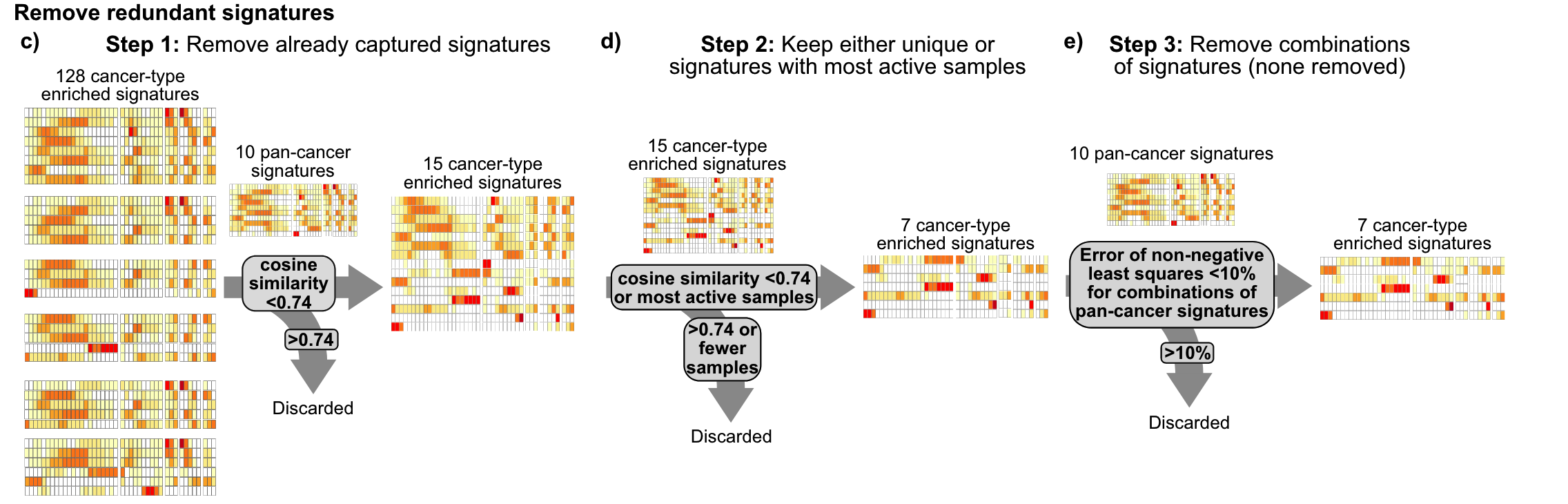
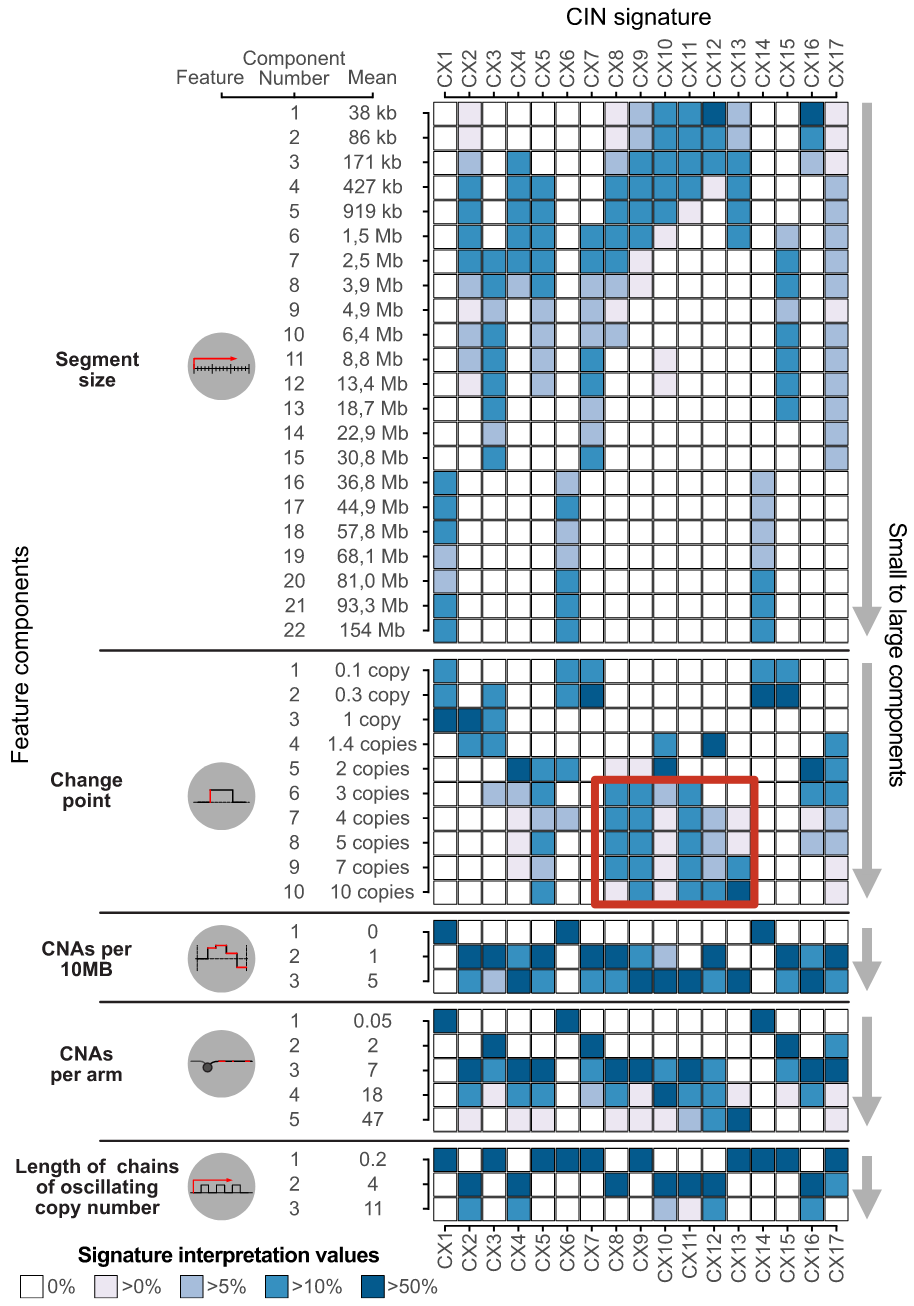
Signature构建
主要结论
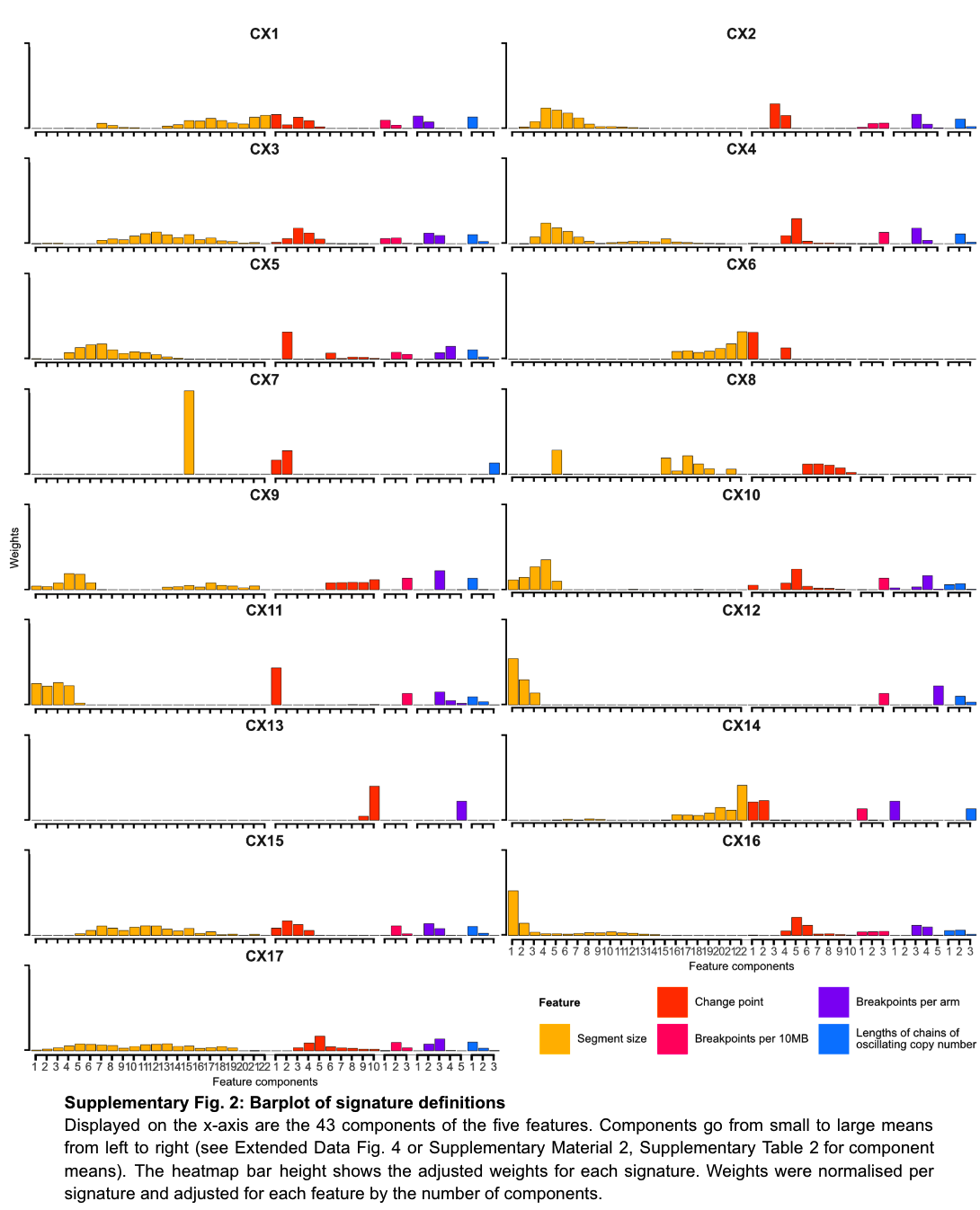
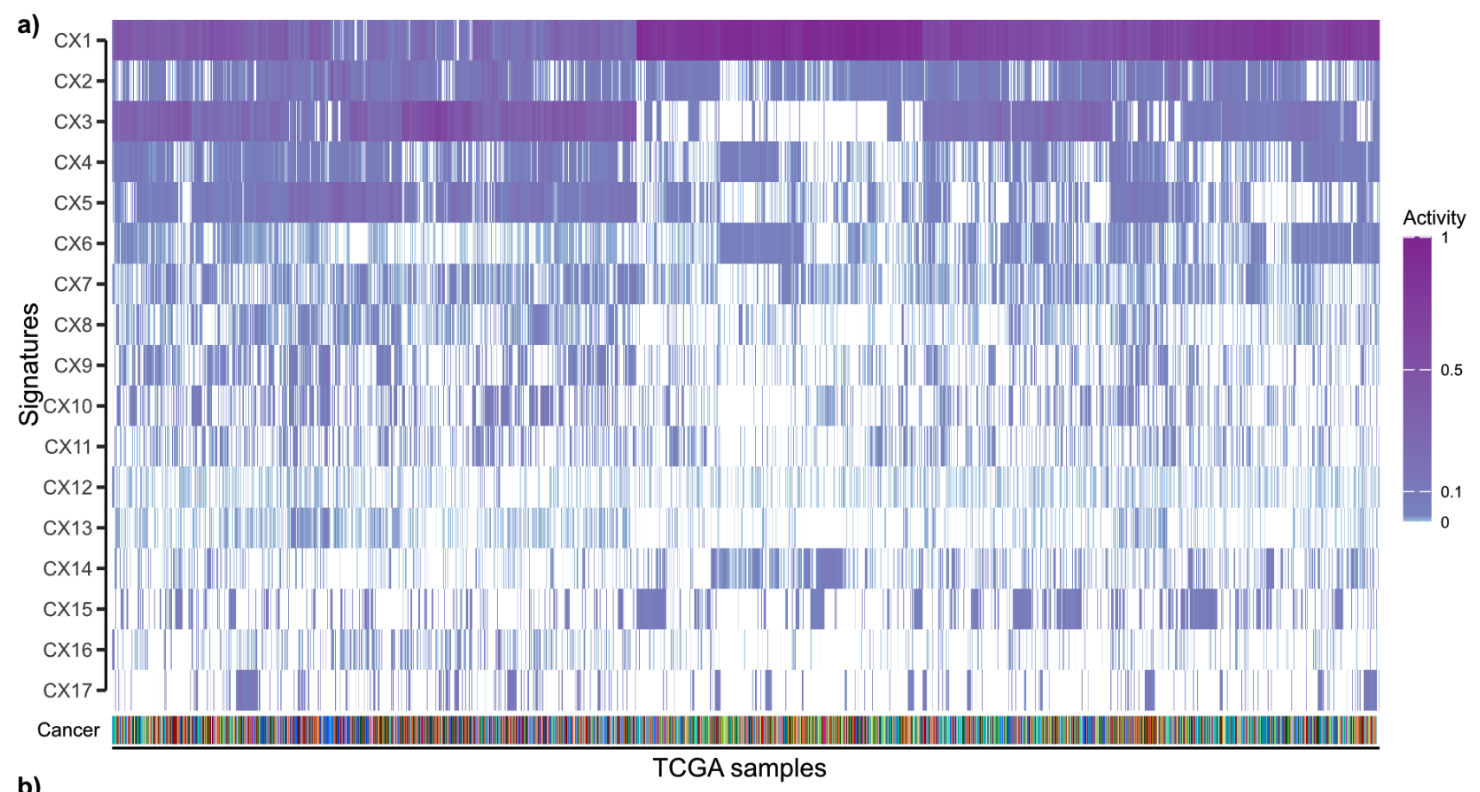
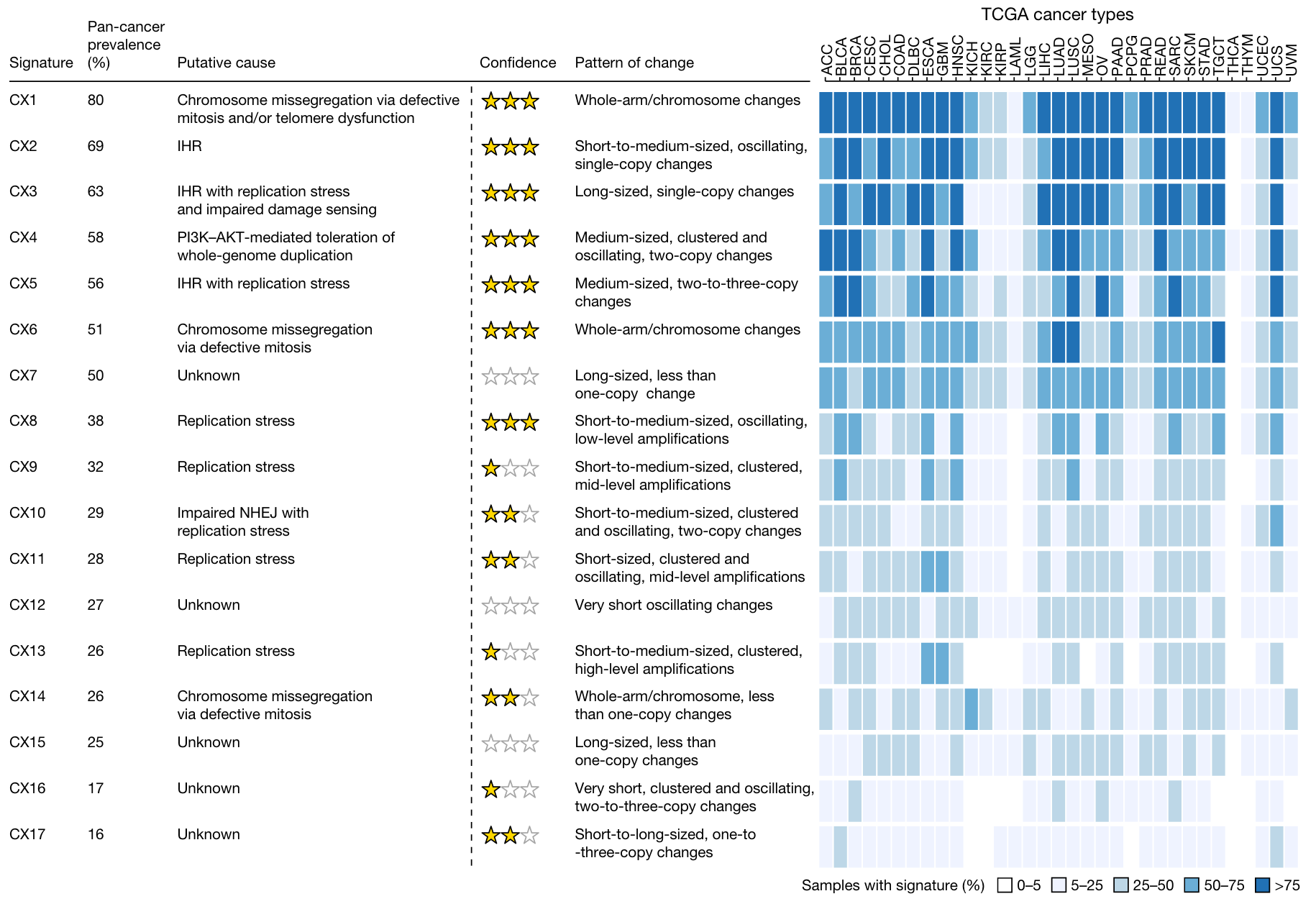
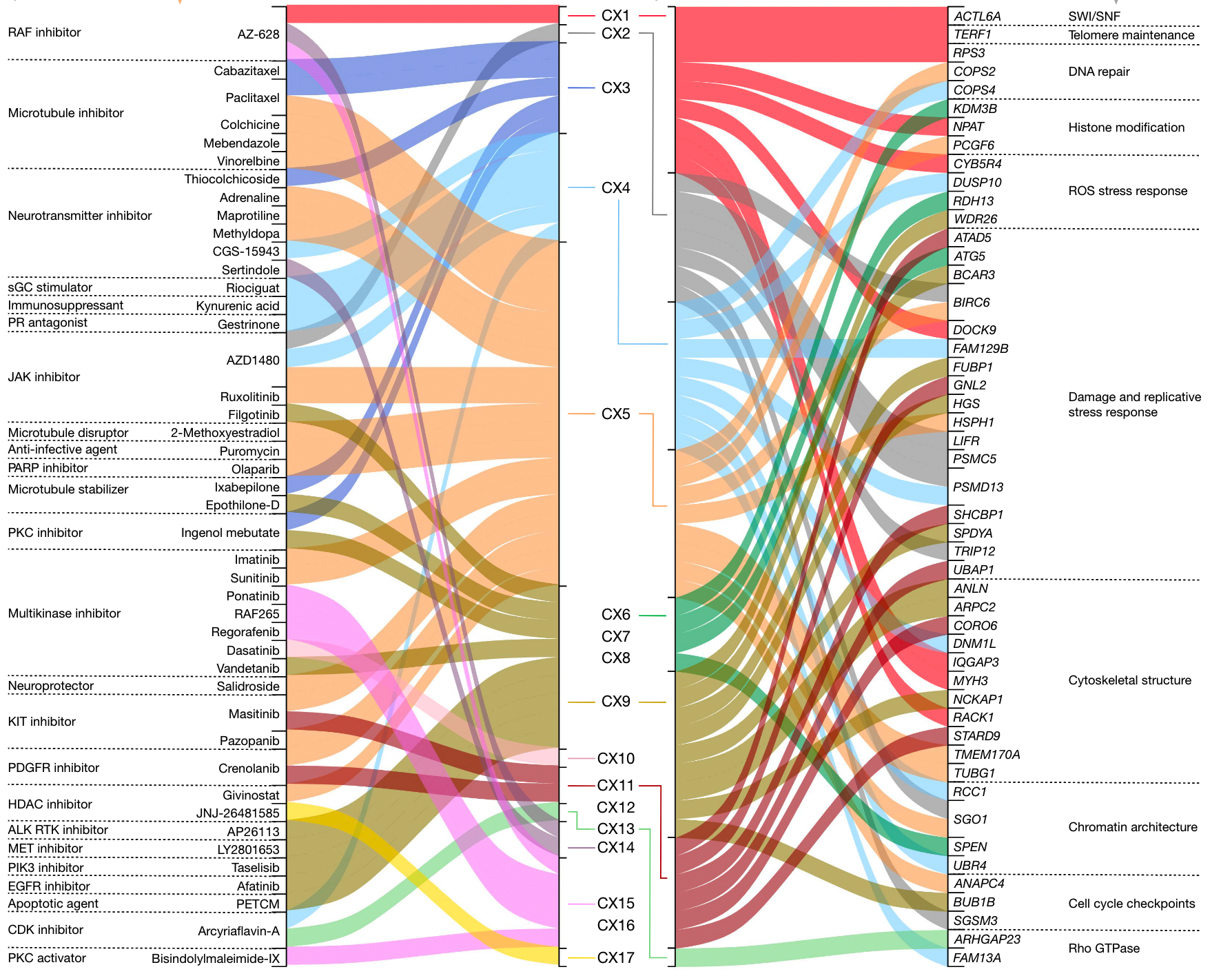
17个拷贝数 signature
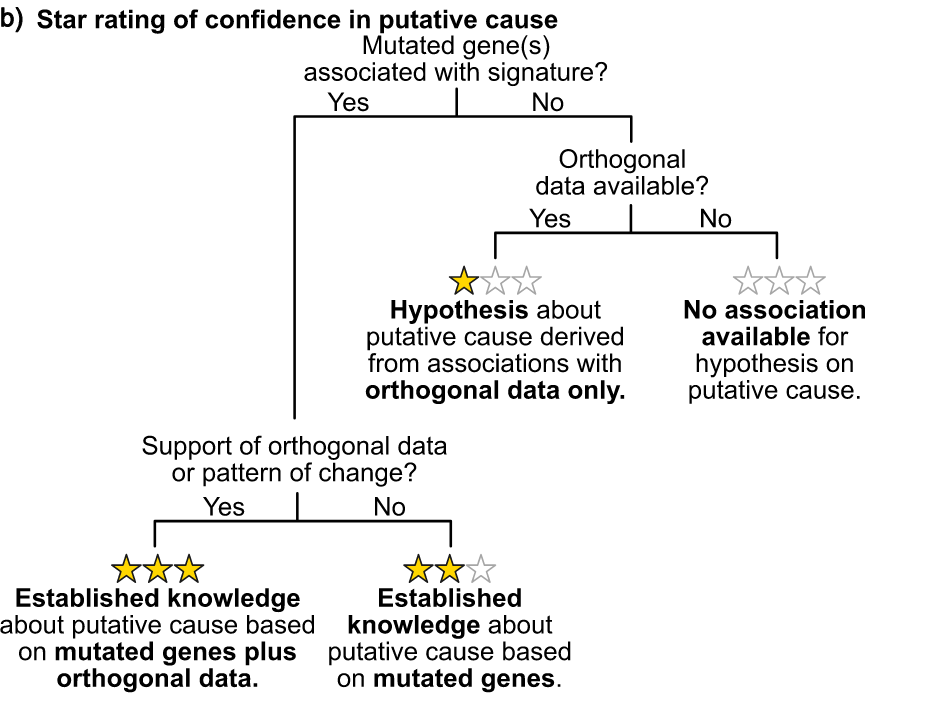
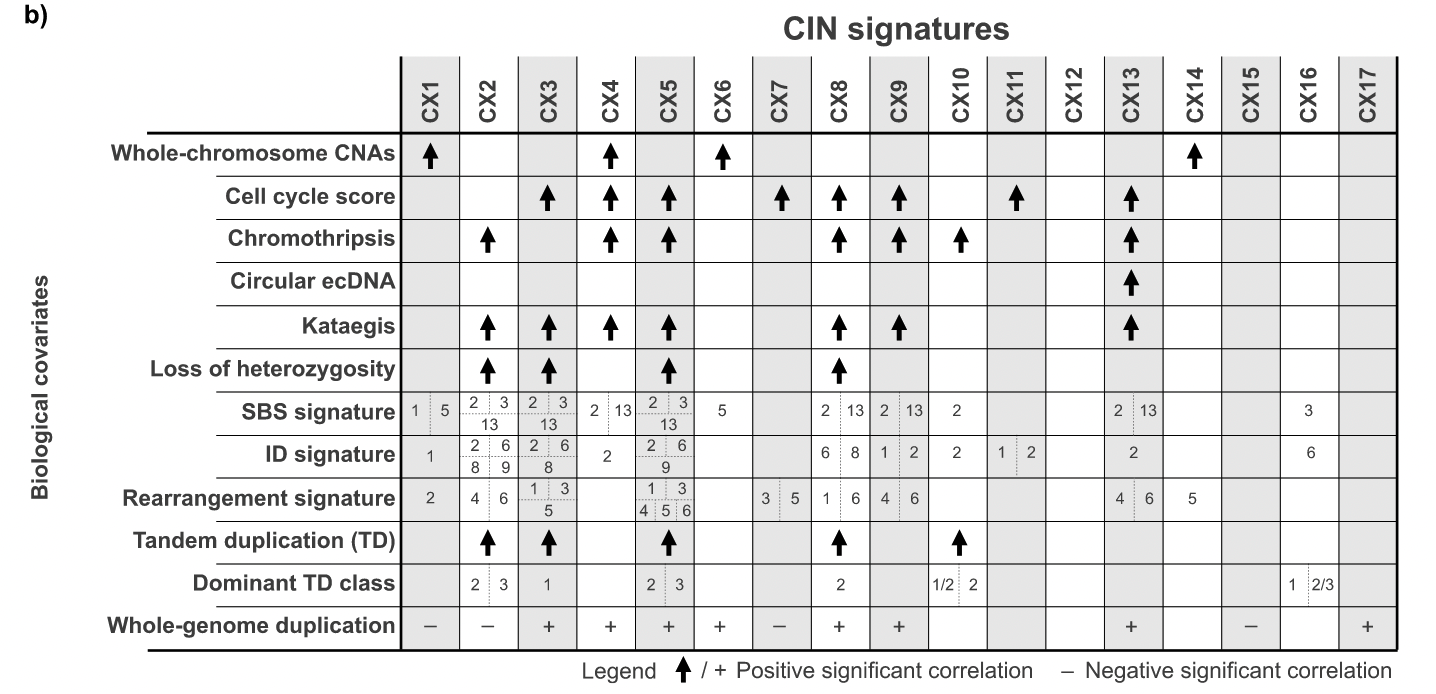
推测不同 signature 成因
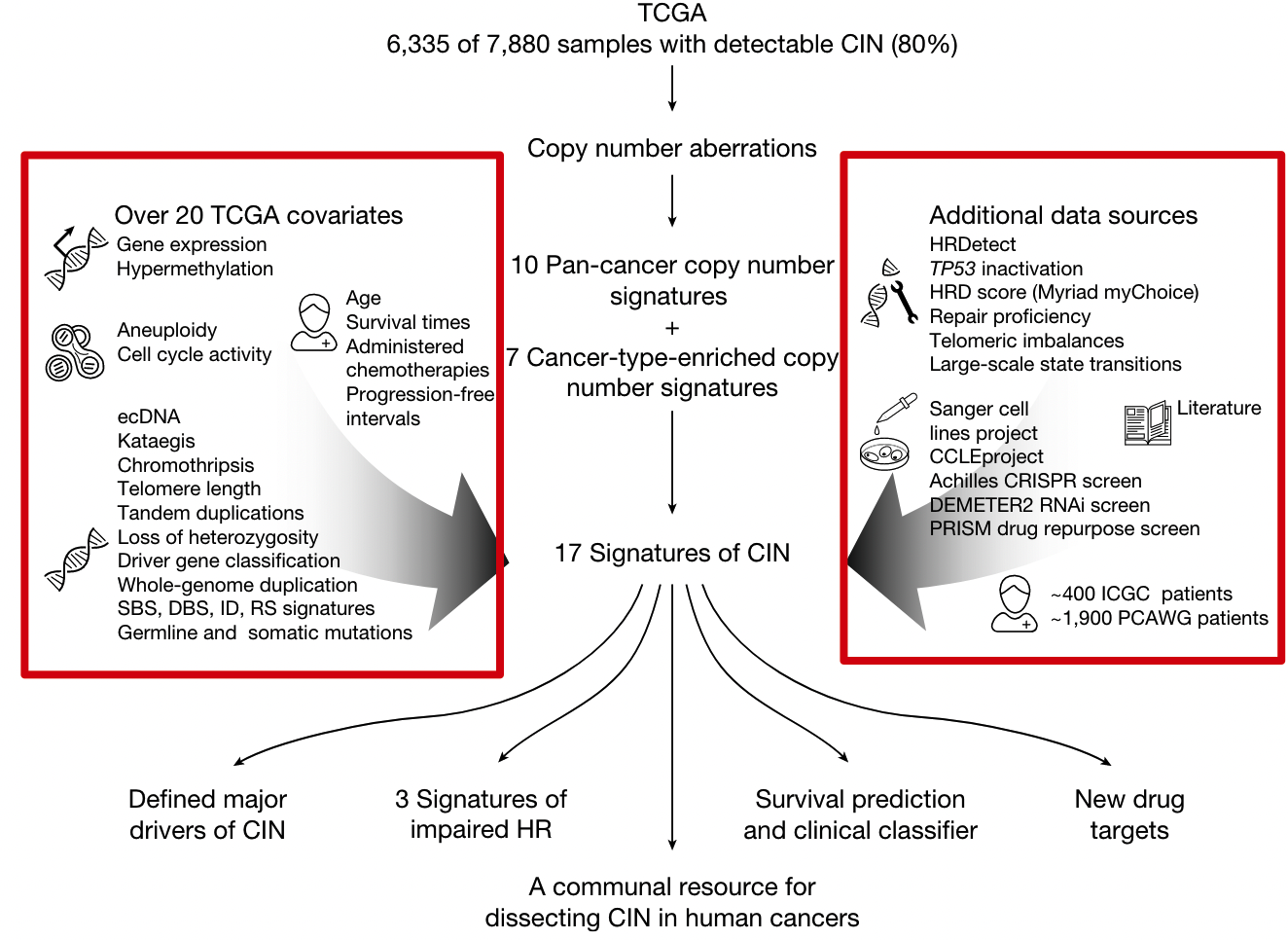
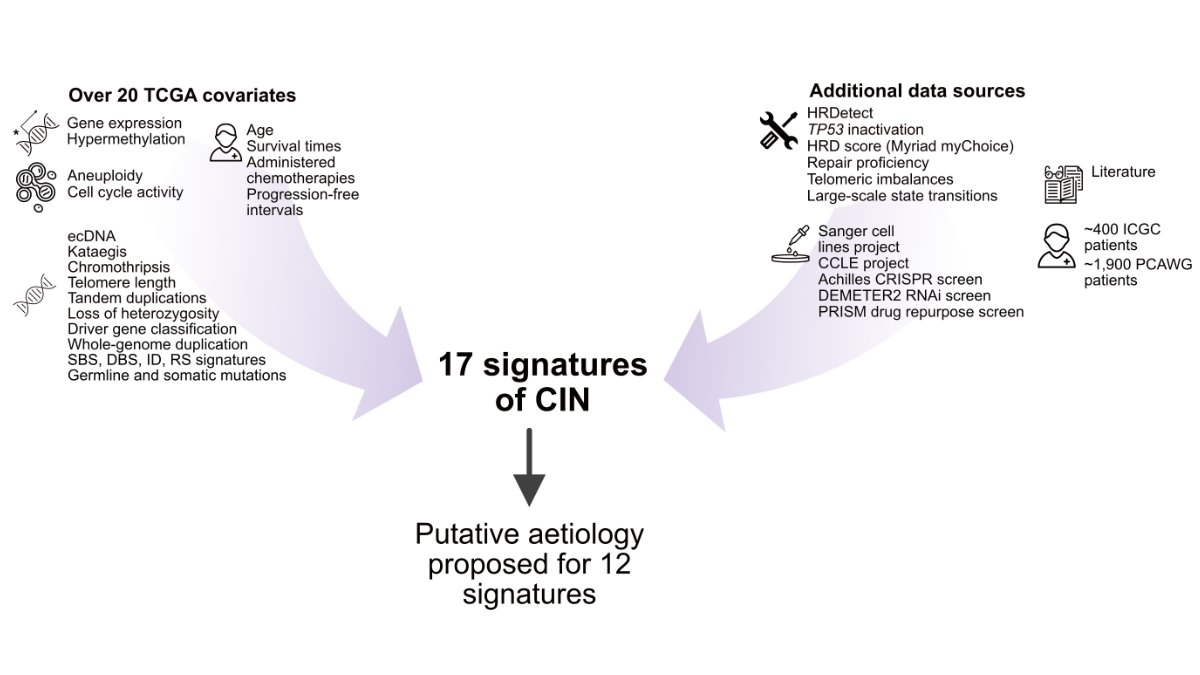
使用数据
重排 rearrangement
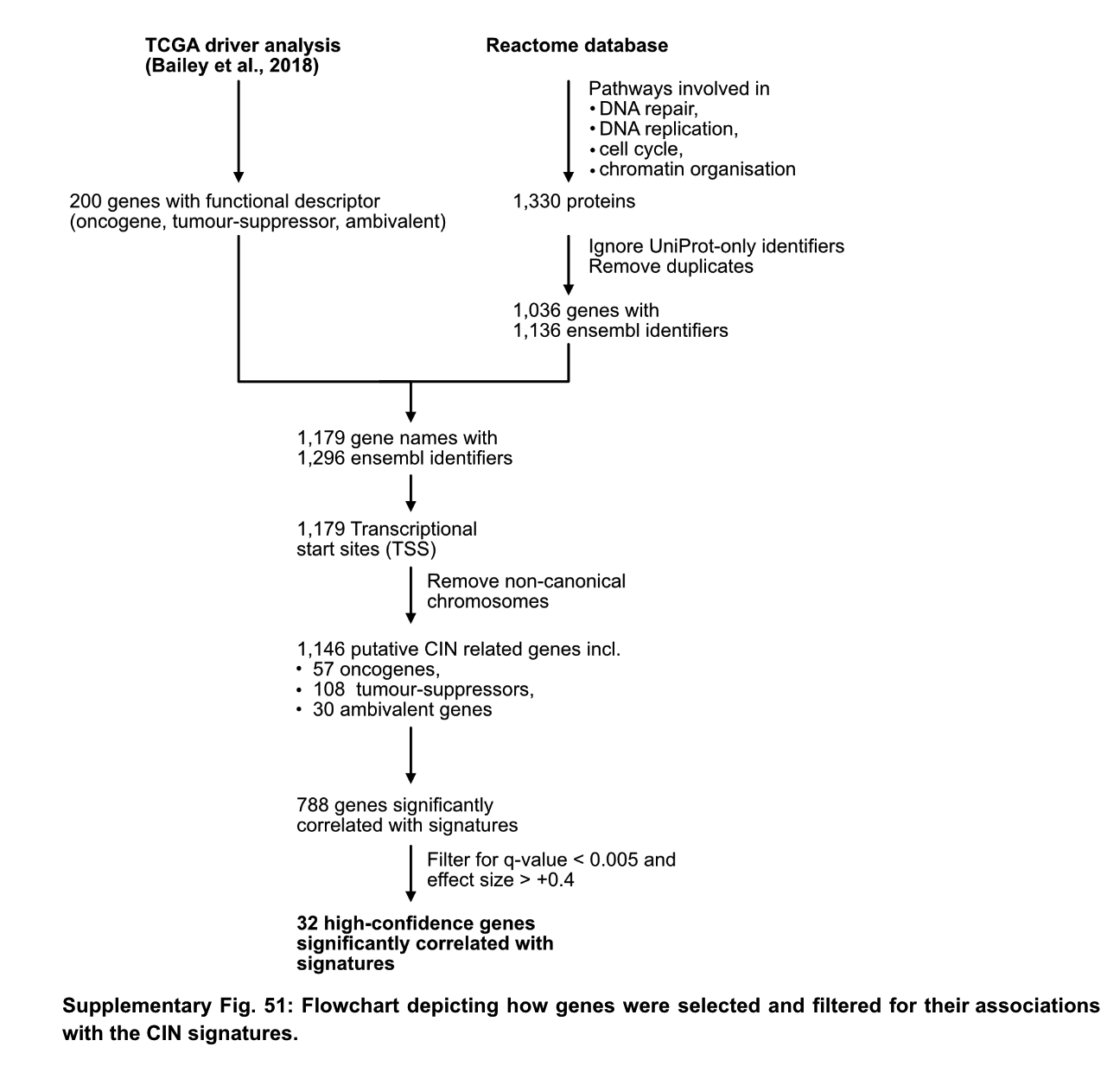
相关CIN基因鉴定
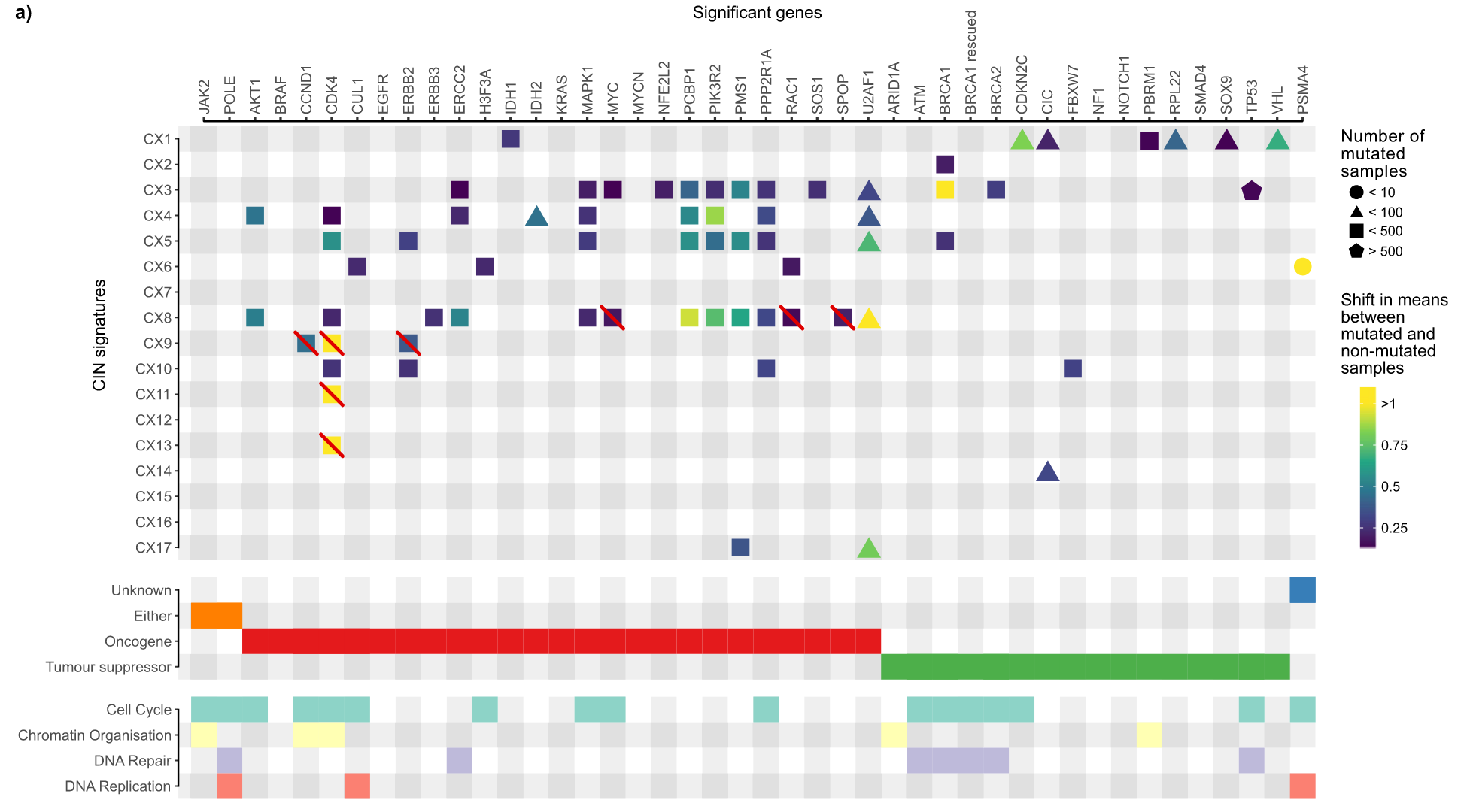
病因假设



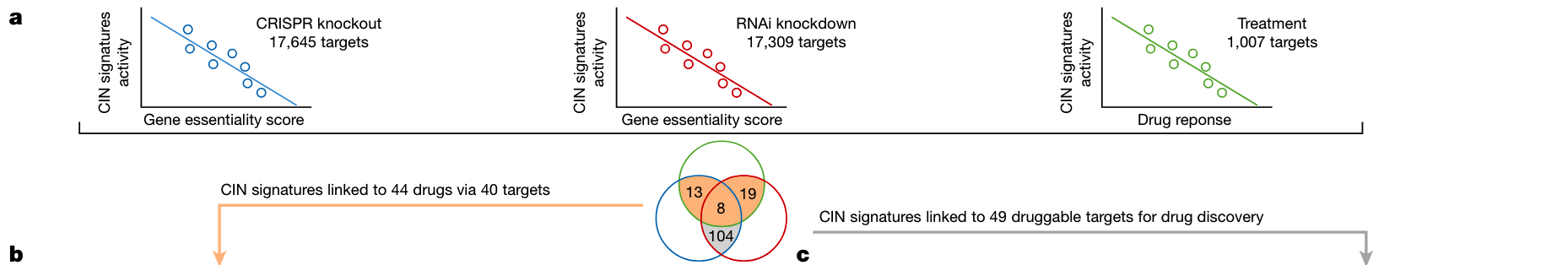
药物反应与药物靶标识别
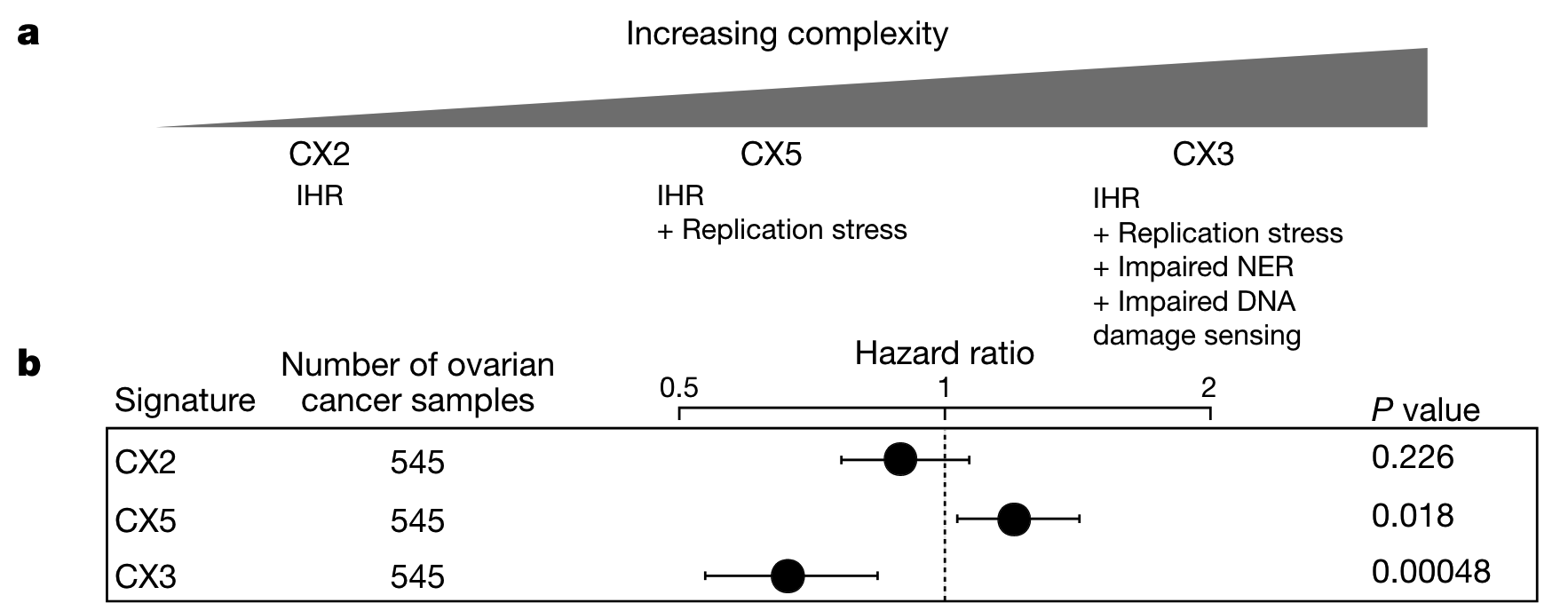
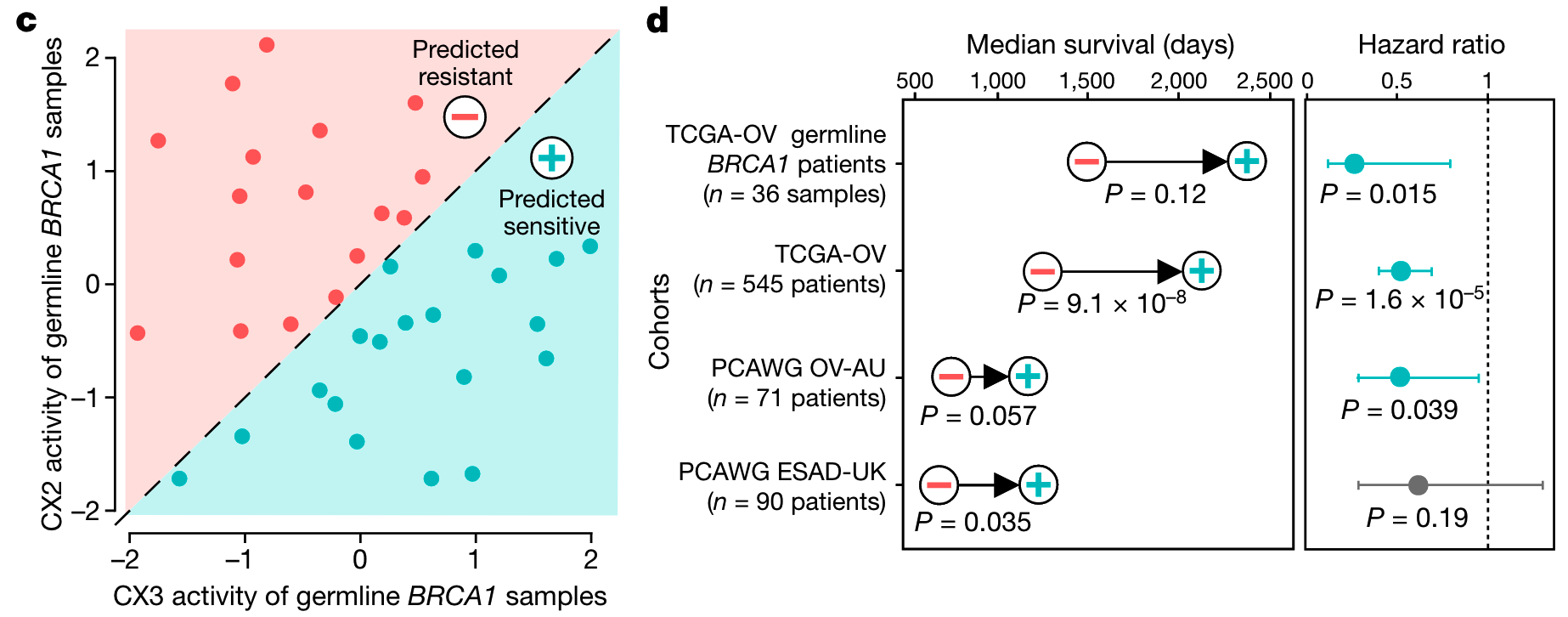
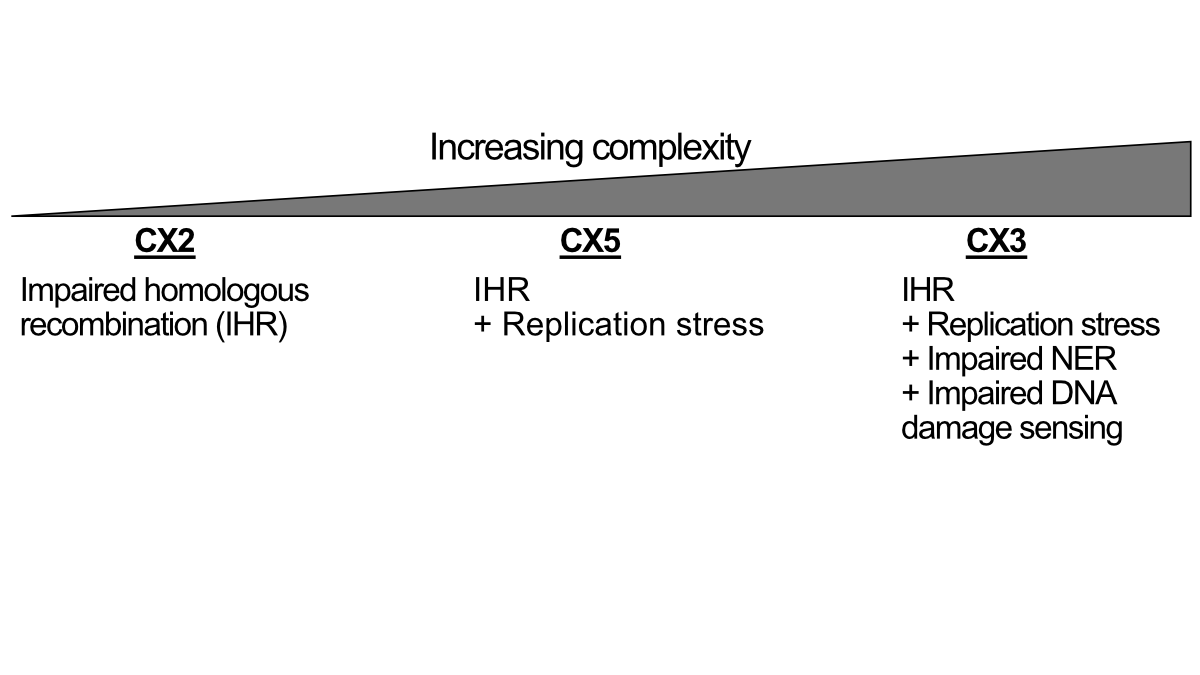
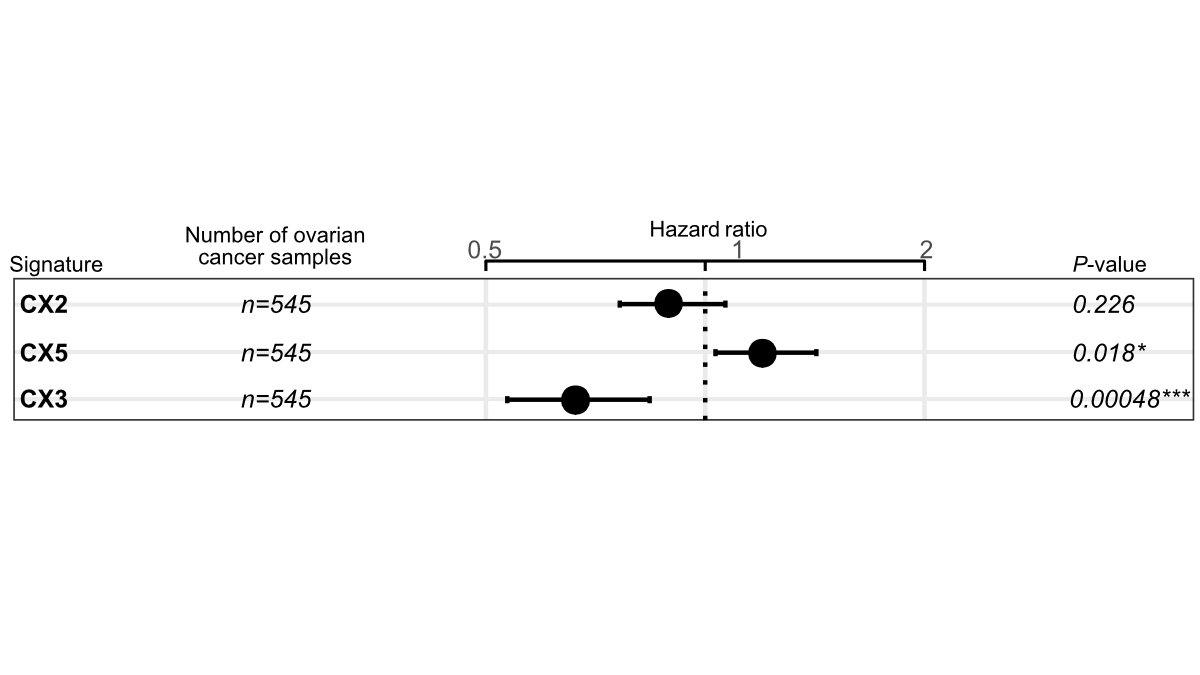
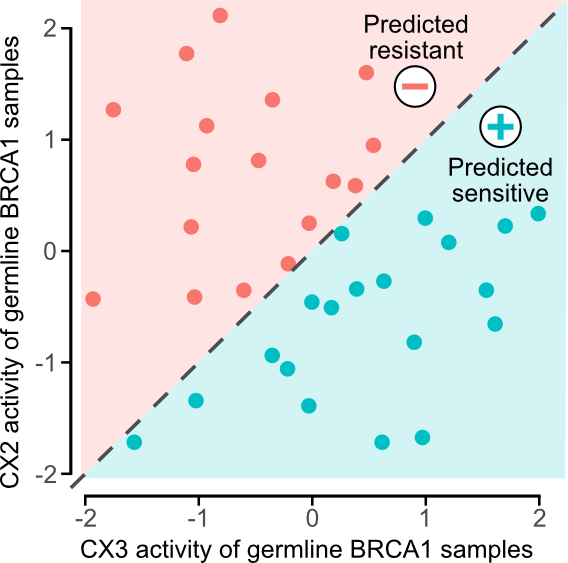
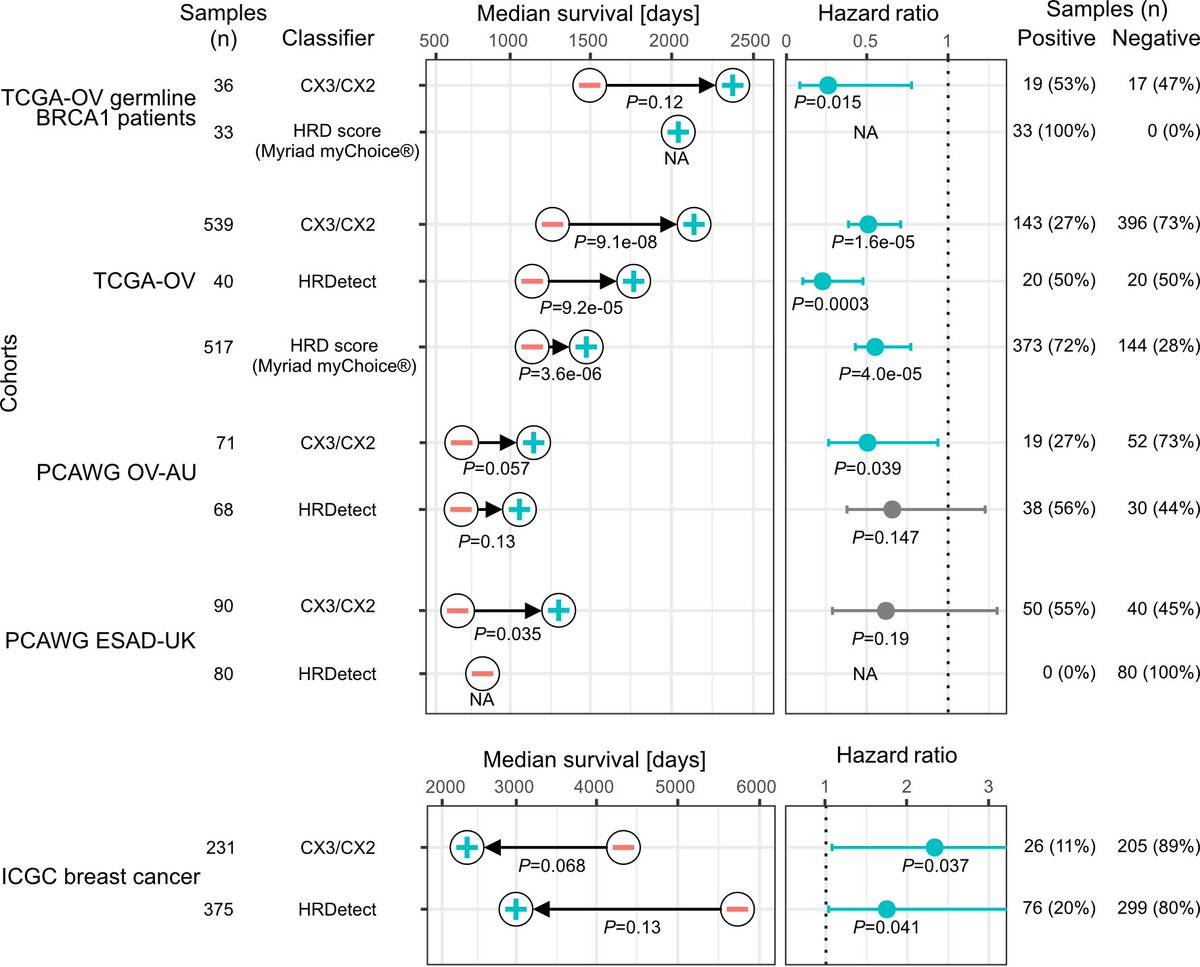
预测铂敏感性
和审稿人反复掰头
第一轮意见

审稿人1
审稿人2

审稿人3

作者更新了什么
第二次意见
审稿人1

审稿人2

审稿人3

作者更新了什么


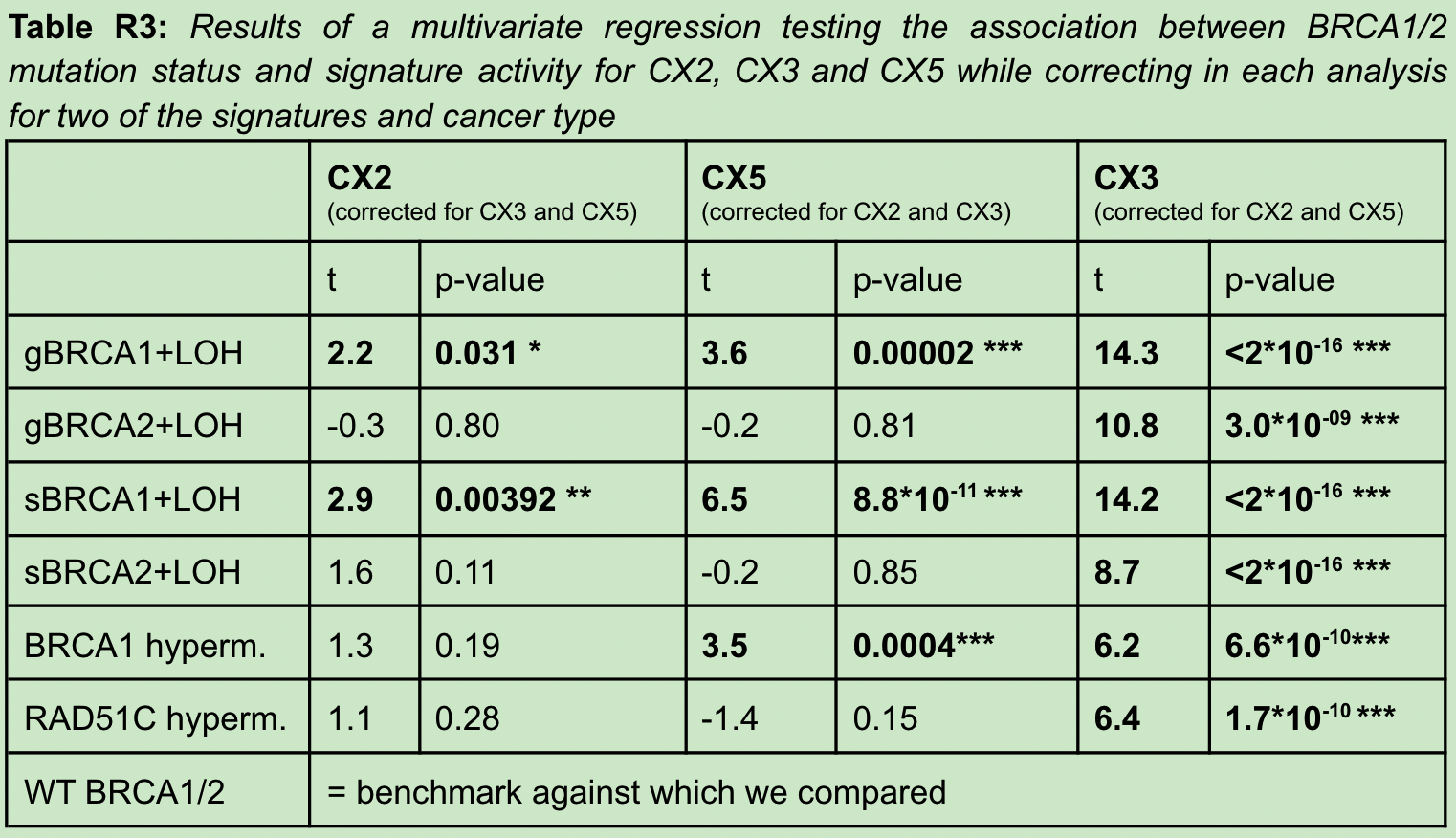

第三次意见


审稿人3

作者更新了什么

第四轮意见

审稿和修改过程
做研究和讲故事
一个好的科学故事

· 分享链接 https://kaopubear.top/blog/2022-06-26-tcga-pancan-cin-signature/